Evo2 在 NPU 上微调的完整流程与问题排查指南
作者:昇腾实战派
背景概述
在生物序列大模型的训练与微调场景中,Evo2 作为基于 Transformer 架构的高性能基因组语言模型,对算力平台提出了较高要求。为实现高效推理与微调,需在昇腾 NPU 环境下完成全流程部署。
本文记录了在Atlas 800I A2平台进行 Evo2 模型微调过程中,从数据准备、预处理到依赖配置与运行时异常的完整处理流程,涵盖常见环境问题与解决方案,为开发者提供可复现的技术参考。
一、数据集下载与合并
原始数据来源于 UCSC 基因组数据库,需下载指定染色体的 FASTA 文件并合并为单个输入文件。
import os
import subprocess
concat_path = "chr17_18_21.fa"
if not os.path.exists(concat_path):
print("文件不存在,开始下载和处理...")
# 创建临时目录
os.makedirs("temp", exist_ok=True)
# 下载文件
files = ["chr17", "chr18", "chr21"]
for f in files:
url = f"https://hgdownload.soe.ucsc.edu/goldenpath/hg38/chromosomes/{f}.fa.gz"
subprocess.run(["wget", url], check=True)
subprocess.run(["zcat", f"{f}.fa.gz", ">", f"{f}.fa"], shell=True)
# 合并文件
with open(concat_path, "w") as out_file:
for f in files:
with open(f"{f}.fa", "r") as in_file:
out_file.write(in_file.read())
print("文件已下载并合并完成")
else:
print("合并后的文件已存在")
⚠️ 若
zcat命令未成功执行,可手动运行,确保文件解压
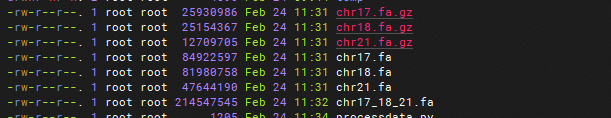
二、数据集预处理配置与转换
使用 processdata.py 生成配置文件,指定输入路径、输出目录及处理参数。
import os
concat_path = "chr17_18_21.fa"
full_fasta_path = os.path.abspath(concat_path)
output_dir = os.path.abspath("preprocessed_data")
output_yaml = f"""
- datapaths: ["{full_fasta_path}"]
output_dir: "/usr/local/Ascend/bionemo-framework"
output_prefix: chr17_18_21_uint8_distinct
train_split: 0.9
valid_split: 0.05
test_split: 0.05
overwrite: True
embed_reverse_complement: true
random_reverse_complement: 0.0
random_lineage_dropout: 0.0
include_sequence_id: false
transcribe: "back_transcribe"
force_uppercase: false
indexed_dataset_dtype: "uint8"
tokenizer_type: "Byte-Level"
vocab_file: null
vocab_size: null
merges_file: null
pretrained_tokenizer_model: null
special_tokens: null
fast_hf_tokenizer: true
append_eod: true
enforce_sample_length: null
ftfy: false
workers: 1
preproc_concurrency: 100000
chunksize: 25
drop_empty_sequences: true
nnn_filter: false # 若按 NNN 分割序列(如人类基因组中的间隙),建议设为 true
seed: 12342 # 无关紧要,因未启用随机反向互补或谱系丢弃
"""
with open("preprocess_config.yaml", "w") as f:
print(output_yaml, file=f)
✅ 生成的
preprocess_config.yaml文件内容如上,确保路径与参数正确。
执行数据预处理命令:
preprocess_evo2 --config preprocess_config.yaml
⚠️ 若命令卡住无响应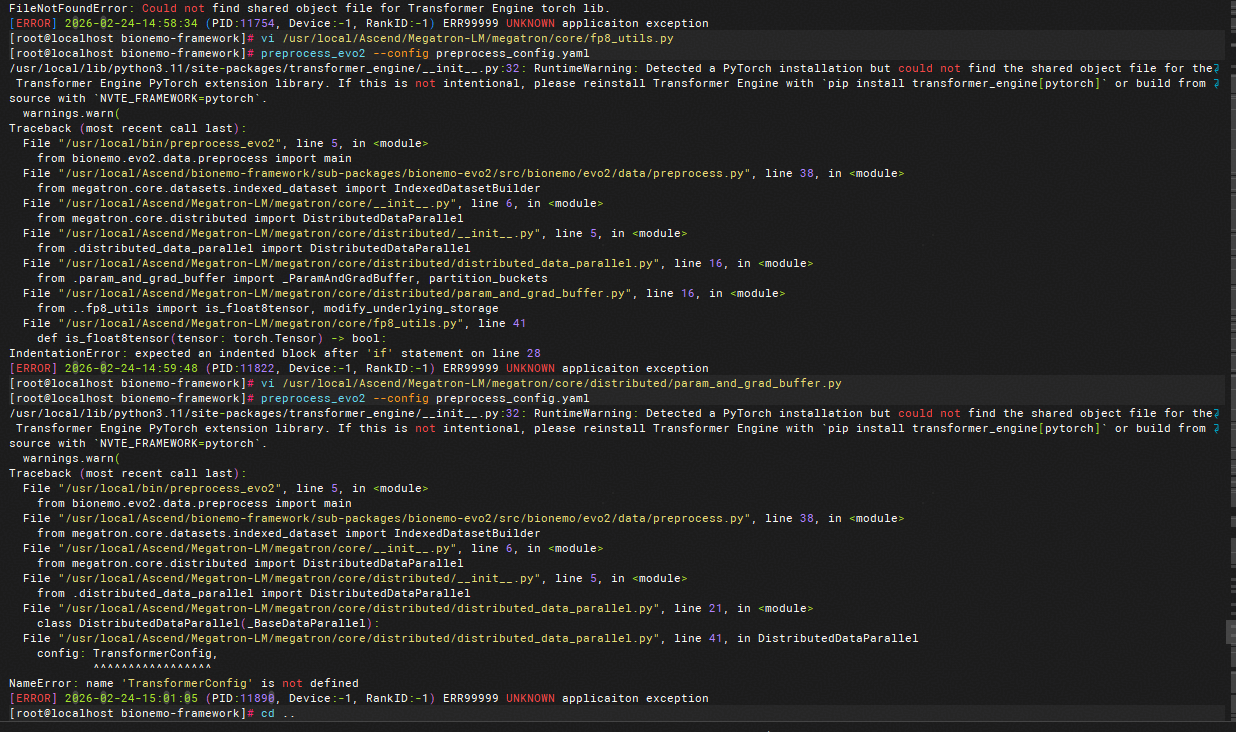
可能为依赖未就绪或环境变量配置缺失,补充一下这个依赖
from mindspeed.core.megatron_basic.arguments_basic import transformer_config_init_wrapper
三、环境依赖与关键模块配置
在执行预处理时,程序进入 mindspeed 模块,提示需引入特定配置类:
from mindspeed.core.megatron_basic.arguments_basic import transformer_config_init_wrapper
✅ 该模块为昇腾生态专用,需确保
Megatron-LM框架已正确安装并加入 Python 路径。
设置环境变量:
export PYTHONPATH=/usr/local/Ascend/Megatron-LM/:$PYTHONPATH
四、Apex 编译与安装
预处理过程中出现 apex 相关报错,需手动构建适配 NPU 的版本。
- 安装基础依赖:
pip install pyyaml
- 克隆并构建 Apex:
git clone https://gitee.com/ascend/apex.git
cd apex/
bash scripts/build.sh --python=3.10
⚠️ 若提示缺少
patch命令,需在具备该工具的环境中执行构建,或通过apt install patch安装。
- 安装生成的 wheel 包:
pip install apex/dist/apex-0.1+ascend-{version}.whl # {version} 替换为实际 Python 版本与 CPU 架构(如 cp310-cp310-linux_x86_64)
✅ 构建成功后,可正常加载
apex模块,解决算子兼容性问题。
五、运行卡顿与镜像依赖问题
尽管依赖已安装,预处理仍可能卡在某处,表现为长时间无输出。
排查发现,部分依赖(如 noodles)在特定环境(如黄区)中无法通过 pip install -e . 完整安装,导致运行时缺失模块。
解决方案:
- 重新构建并传输完整镜像,确保所有依赖项(包括
noodles、mindspeed、bionemo-framework)均在镜像中预置。 - 或在目标环境中逐项安装缺失依赖,使用
pip install -e <path>确保本地开发包正确加载。
六、总结与建议
- 数据准备阶段:确保 FASTA 文件完整解压,路径正确。
- 配置文件:使用绝对路径,避免相对路径导致路径解析错误。
- 环境变量:
PYTHONPATH必须包含Megatron-LM及mindspeed路径。 - Apex 构建:需在支持
patch的环境中构建,避免编译失败。 - 依赖完整性:优先使用完整镜像部署,避免因依赖缺失导致运行卡顿。
通过以上步骤,可顺利完成 Evo2 在 NPU 上的数据预处理与微调环境搭建,为后续模型训练与推理提供稳定支持。

AtomGit 是由开放原子开源基金会联合 CSDN 等生态伙伴共同推出的新一代开源与人工智能协作平台。平台坚持“开放、中立、公益”的理念,把代码托管、模型共享、数据集托管、智能体开发体验和算力服务整合在一起,为开发者提供从开发、训练到部署的一站式体验。
更多推荐



 2
2 0
0- 0



 已为社区贡献1条内容
已为社区贡献1条内容

所有评论(0)