TensorQTL介绍+实操
eQTL概述
eQTL (expression Quantitative Trait Loci,表达数量性状位点) 是基因组中那些能够影响基因表达水平的特定变异位点(通常是 SNP)。
eQTL 分析的本质
是在检验 n 个样本中,基因型 X(自变量)和基因表达量 Y(因变量)之间是否存在相关性。
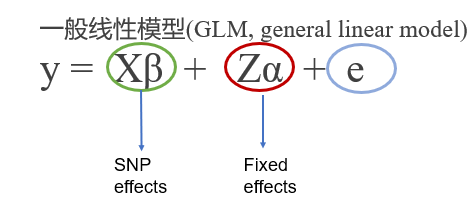
什么是tensorQTL?
tensorQTL 是一个基于 PyTorch 的 Python 软件库,专为大规模分子 QTL 映射设计。通过利用 GPU 的大规模并行计算能力,它在执行顺式(cis)和反式(trans)QTL 映射时,速度较传统的 FastQTL 和 MatrixQTL 有了质的飞跃,特别是在置换检验(Permutation test)中,性能提升可达数百倍。
TensorQTL的主要运用场景
tensorQTL主要用于分析基因型(Genotypes)与表型(Phenotypes)之间的关联。在eQTL里面表型指基因表达量。
最常见的包括:
cis-QTL映射:寻找物理距离上靠近基因的变异位点(SNP)如何影响该基因的表达
trans-eQTL映射:寻找远距离变异位点对基因的影响(计算量较大)
sQTL:分析变异如何影响RNA的剪接模式
TensorQTL实操:
官方指南:https://github.com/broadinstitute/tensorqtl
1. 环境搭建
#创建环境
conda activate tensorqtl_env python=3.9
#安装tensorqtl
pip3 install tensoratl
#安装最新版本
pip install pip@git+https://github.com/broadinstitute/tensorqtl.git
#如果要使用PLINK2二进制文件(pgen/pvar/psam),需要安装pgenlib
pip install Pgenlib2. 环境检查与路径定义
文件地址:https://github.com/broadinstitute/tensorqtl/tree/master/example/data
import pandas as pd
import torch
import tensorqtl
import os
from tensorqtl import pgen, cis, trans, post
#define paths to data
plink_prefix_path = 'GEUVADIS.445_samples.GRCh38.20170504.maf01.filtered.nodup.chr18'
expression_bed = 'GEUVADIS.445_samples.expression.bed.gz'
covariates_file = 'GEUVADIS.445_samples.covariates.txt'
prefix = 'GEUVADIS.445_samples'
3. 加载表型与协变量
# load phenotypes and covariates
print(">> Loading phenotypes and covaruates")
phenotype_df, phenotype_pos_df = tensorqtl.read_phenotype_bed(expression_bed)
covariates_df = pd.read_csv(covariates_file, sep='\t', index_col=0).T
4. 加载基因型
# PLINK reader for genotypes
print("加载基因型")
pgr = pgen.PgenReader(plink_prefix_path)
genotype_df = pgr.load_genotypes()
variant_df = pgr.variant_df
# map all cis-associations (results for each chromosome are written to file)
5. 核心分析逻辑
①cis_nominal mapping
# all genes
# cis.map_nominal(genotype_df, variant_df, phenotype_df, phenotype_pos_df, prefix, covariates_df=covariates_df)
# genes on chr18
print("cis")
cis.map_nominal(genotype_df, variant_df,
phenotype_df.loc[phenotype_pos_df['chr'] == 'chr18'],
phenotype_pos_df.loc[phenotype_pos_df['chr'] == 'chr18'],
prefix, covariates_df=covariates_df)
②cis.map_cis permutation
#cis-QTL:empirical p-values for phenotypes
print("cis-QTL:empirical")
cis_df = cis.map_cis(genotype_df, variant_df,
phenotype_df.loc[phenotype_pos_df['chr'] == 'chr18'],
phenotype_pos_df.loc[phenotype_pos_df['chr'] == 'chr18'],
covariates_df=covariates_df,seed=123456)
6. 后处理与保存
①后处理
#compute q-values(in pratice, this must be run on all genes, not a subset)
post.calculate_qvalues(cis_df,fdr=0.05,qvalue_lambda=0.85)
②保存
print(cis_df.columns.tolist())
output_dir = "results"
os.makedirs(output_dir, exist_ok=True)
final_out_file = os.path.join(output_dir, f"{prefix}.cis_qtl_chr18_w_permutation.txt.gz")
print(f"Saving results to: {final_out_file}")
cis_df.to_csv(
final_out_file,
sep='\t',
index=False,
compression='gzip',
float_format='%.8g',
na_rep='NA'
)
7. trans mapping
trans_df = trans.map_trans(genotype_df, phenotype_df, convariates_df, batch_size=20000,
return_sparse = Ture, pval_threshold=1e-5, maf_threshold=0.05)
# remove cis-associations
trans_df = trans.filter_cis(trans_df, phenotype_pos_df.T.to_dict(), variant_df, window=5000000)结果展示
Norminal mapping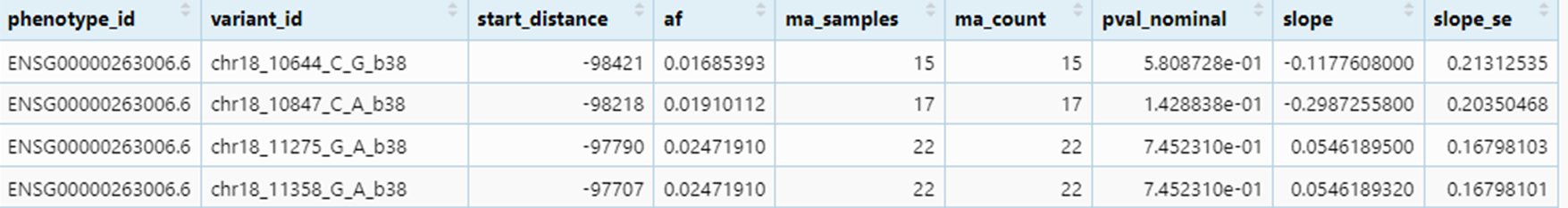
|
列名 |
解释 |
|
phenotype_id |
表型ID(通常是基因ID) |
|
variant_id |
变异ID(SNP ID);PS:b38,指组装版本 |
|
start_distance |
到转录起始位点(TSS)的距离 |
|
af |
等位基因频率(通常指次要等位基因频率 MAF) |
|
ma_samples |
携带次要等位基因的样本数 |
|
ma_count |
次要等位基因计数 |
|
pval_nominal |
名义 p 值(原始 p 值,未校正) |
|
slope |
效应大小(回归系数 β) |
|
slope_se |
slope 的标准误(standard error) |
Permutation mapping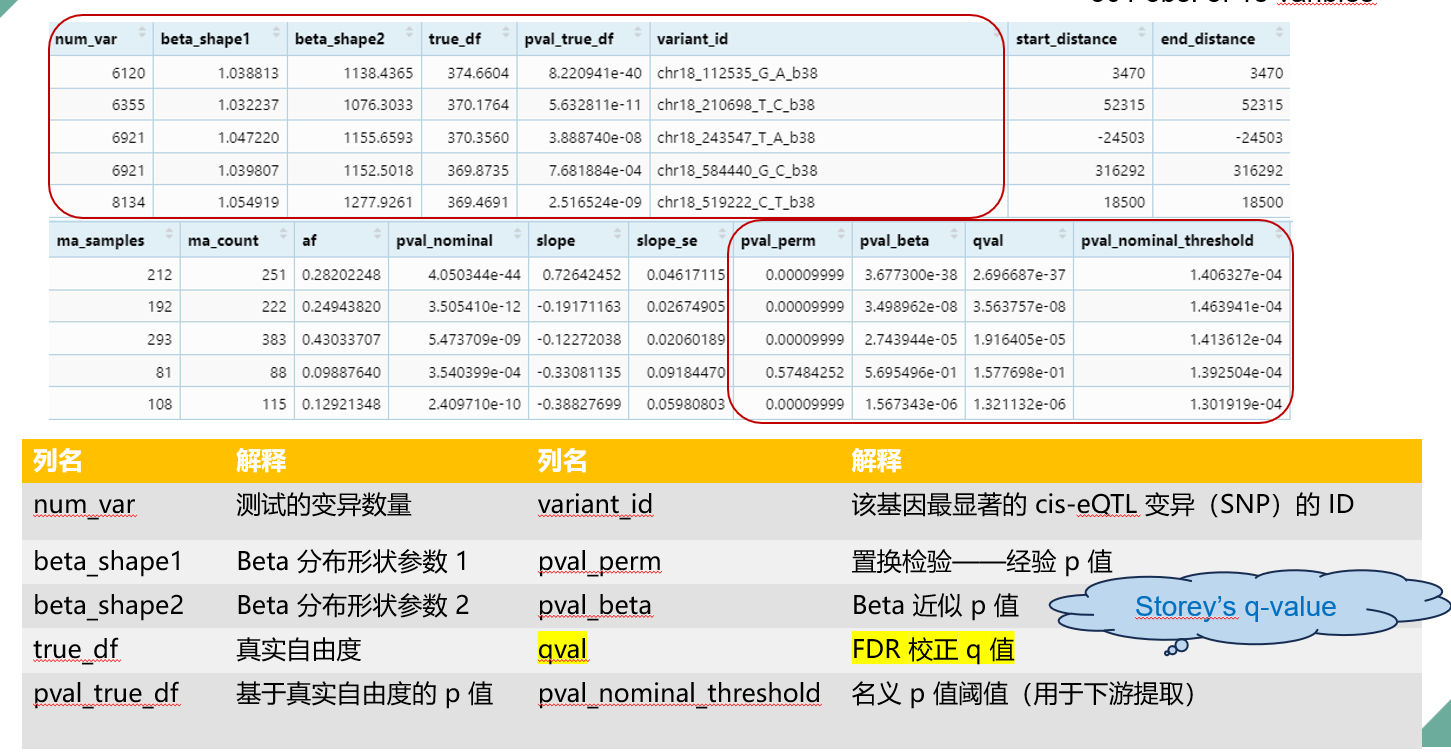
trans mapping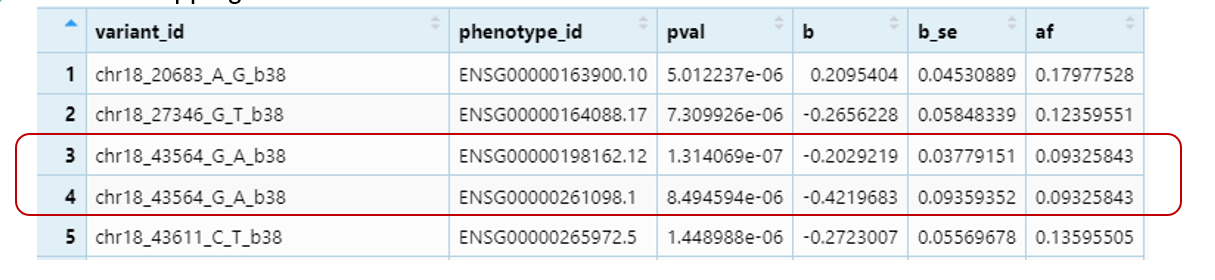
红色框框内,可以看到,变异SNP的ID一致,但却map到了不同的基因,暗示该SNP可能具有多效性。

AtomGit 是由开放原子开源基金会联合 CSDN 等生态伙伴共同推出的新一代开源与人工智能协作平台。平台坚持“开放、中立、公益”的理念,把代码托管、模型共享、数据集托管、智能体开发体验和算力服务整合在一起,为开发者提供从开发、训练到部署的一站式体验。
更多推荐

 6
6 0
0- 0



 已为社区贡献1条内容
已为社区贡献1条内容

所有评论(0)