如何用RNA速率“倒放“细胞命运的演化录像?
单细胞RNA测序(scRNA-seq)就像给细胞群体拍了一张高清合影——你能看清每个细胞的"表情",却猜不透它们下一秒要"做什么"。
如果告诉你,仅凭一张静态照片,我们就能推算出细胞过去的状态和未来的命运,甚至预测它正在分化还是即将凋亡——这听起来像科幻,但RNA速率(RNA Velocity) 正是这样一台"分子时间机器"。
什么是RNA速率?
想象基因表达是一场接力赛:
-
未剪接mRNA(unspliced) = 刚接过棒、还在热身的选手(新生转录本)
-
剪接mRNA(spliced) = 正在全力冲刺的选手(成熟转录本)
通过追踪这两种状态的"人数比例",我们不仅能知道现在谁领先,还能推算出接力棒传递的速度和方向——这就是RNA速率的本质:利用剪接动力学模型,从静态测序数据中重建细胞状态的时间导数(变化速率)。
关键洞察:未剪接与剪接mRNA的比率如同细胞的"代谢指纹"——比率升高暗示该基因正在被激活(诱导),比率下降则预示表达抑制。这种动态信息让我们能够:
-
🧭 推断轨迹方向:细胞是走向分化终点,还是停留在稳定状态?
-
⏱️ 预测伪时间:在发育路径上为每个细胞精确"定位"
-
🎯 识别驱动基因:找到推动命运转变的关键调控因
PS:本篇文章是针对百创智造S3000空间转录组技术来展开如何进行RNA速率分析的
分析步骤:
原始测序数据 (BAM)
↓
【Step 1】bam文件添加分子标签 → 区分剪接状态
↓
【Step 2】Velocyto处理 → 生成.loom动态矩阵
↓
【Step 3】R/Scanpy分析 → 速率嵌入与可视化
↓
🎁 产出:细胞命运预测的"动态地图"环境配置:
#R中需要安装的包
install.packages('Matrix','velocyto.R','Seurat','SeuratWrappers','tidyverse')
conda install numpy scipy cython numba matplotlib scikit-learn h5py click
pip install velocyto
一、 将bam文件添加tag
给level水平的bam文件添加tag的脚本:bam_add_flag.pl
通过网盘分享的文件:细胞分割的bam文件添加细胞ID
链接: https://pan.baidu.com/s/1SAxfvi9gNIJcZJW-K4hWSQ 提取码: e438
参数说明
perl bam_add_flag.pl <bc_umi_read.tsv.id> <BSTViewer_project/subdata/L1_heAuto/barcodes.tsv.gz> <sample/barcode.tsv> <BSTViewer_project/level_matrix/level_1/barcodes_cluster.tsv.gz> <in.bam> <out.bam>
#shell中运行,将bam文件添加tag
perl /path/to/bam_add_flag.pl
/share/nas1/sunh/project/demo/out1/01.fastq2BcUmi/S3000-SN-DEMO.bc_umi_read.tsv.id \
/share/nas1/sunh/project/demo/out1/05.AllheStat/BSTViewer_project/subdata/L18_heAuto/barcodes.tsv.gz \
/share/nas1/sunh/project/demo/out1/S3000-SN-DEMO/barcode.tsv \
/share/nas1/sunh/project/demo/out1/05.AllheStat/BSTViewer_project/level_matrix/level_18/barcodes_cluster.tsv.gz \
/share/nas1/sunh/project/demo/out1/02.Umi2Gene/S3000-SN-DEMOAligned.out.bam \ #bam文件在02.Umi2Gene目录
/share/nas1/sunh/project/demo/out1/analysis/level18/new_out.bam L18
#shell中运行
#将bam文件按照位置进行排序
samtool sort /share/nas1/sunh/project/demo/out1/analysis/level18/new_out.bam -o /share/nas1/sunh/project/demo/out1/analysis/level18/new_out.sort.bam
#将排序后的bam构建索引
samtools index /share/nas1/sunh/project/demo/out1/analysis/level18/new_out.sort.bam
二、velocyto生成loom文件
velocyto github网站:https://github.com/velocyto-team/velocyto.py
velocyto文档:https://velocyto.org/velocyto.py
#shell
nohup velocyto run \
-o /share/nas1/sunh/project/demo/whole_out/analysis/level/velocyto/out \ #此为loom结果输出目录
/share/nas1/sunh/project/demo/out1/analysis/level18/new_out.sort.bam \ #此为排序后的bam文件
/share/nas1/dengdj/testing/Gene_expression/ref_GRCm38_release95/genes/genes.gtf -@ 10三、在R中运行velocyto.R
#在R中运行
#转化loom文件为h5ad文件
library(Matrix)
library(velocyto.R)
library(Seurat)
library(SeuratWrappers)
library(tidyverse)
velo <- ReadVelocity(file = "/share/nas1/sunh/project/demo/whole_out/analysis/level/velocyto/out/chr12_3MYNC.loom")
rownames(velo$spliced) <- make.unique(rownames(velo$spliced))
rownames(velo$unspliced) <- make.unique(rownames(velo$unspliced))
rownames(velo$ambiguous) <- make.unique(rownames(velo$ambiguous))
velo <- as.Seurat(x = velo)
velo <- velo %>% SCTransform(assay="spliced") %>% RunPCA(verbose=F)
ElbowPlot(velo, ndims = 50)
nPC=1:20
velo <- FindNeighbors(velo, dims = nPC) %>% FindClusters() %>%
RunUMAP(dims = nPC) %>% RunTSNE(dims = nPC)
##给细胞分配颜色
ident.colors <- (scales::hue_pal())(n = length(x = levels(x = velo)))
names(x = ident.colors) <- levels(x = velo)
cell.colors <- ident.colors[Idents(object = velo)]
names(x = cell.colors) <- colnames(x = velo)
##速率分析
velo <- RunVelocity(velo, deltaT = 1, kCells = 25, fit.quantile = 0.02,
spliced.average = 0.2, unspliced.average = 0.05, ncores = 18)
#kCells:用于斜率平滑度计算最近邻细胞数量,越大越平滑,越小越能反映局部特征
#fit.quantile:0.02代表对基因表达量最高2%与最低2%的值执行gamma拟合
#spliced.average:过滤低表达丰度基因的标准,计算的是基因在cluster内的平均counts值
#unspliced.average:同上
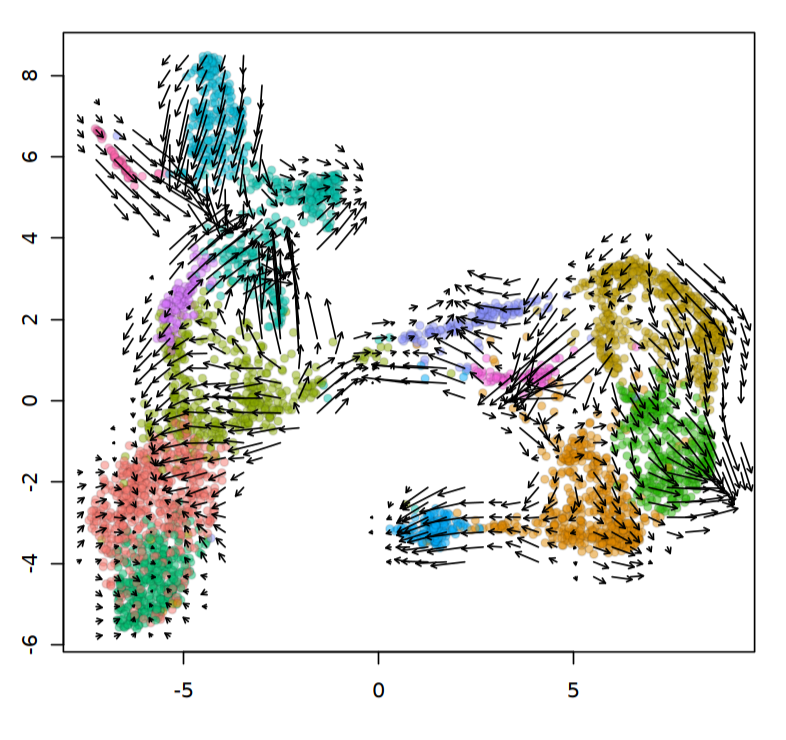
全局速率可视化
DimPlot(velo, reduction = "umap")
emb = Embeddings(velo, reduction = "umap")
vel = Tool(velo, slot = "RunVelocity")
show.velocity.on.embedding.cor(emb = emb, vel = vel, n = 200, scale = "sqrt",
cell.colors = ac(cell.colors, alpha = 0.5), cex = 0.8, arrow.scale = 3,
show.grid.flow = TRUE, min.grid.cell.mass = 0.5, grid.n = 40,
arrow.lwd = 1, do.par = FALSE, cell.border.alpha = 0.1)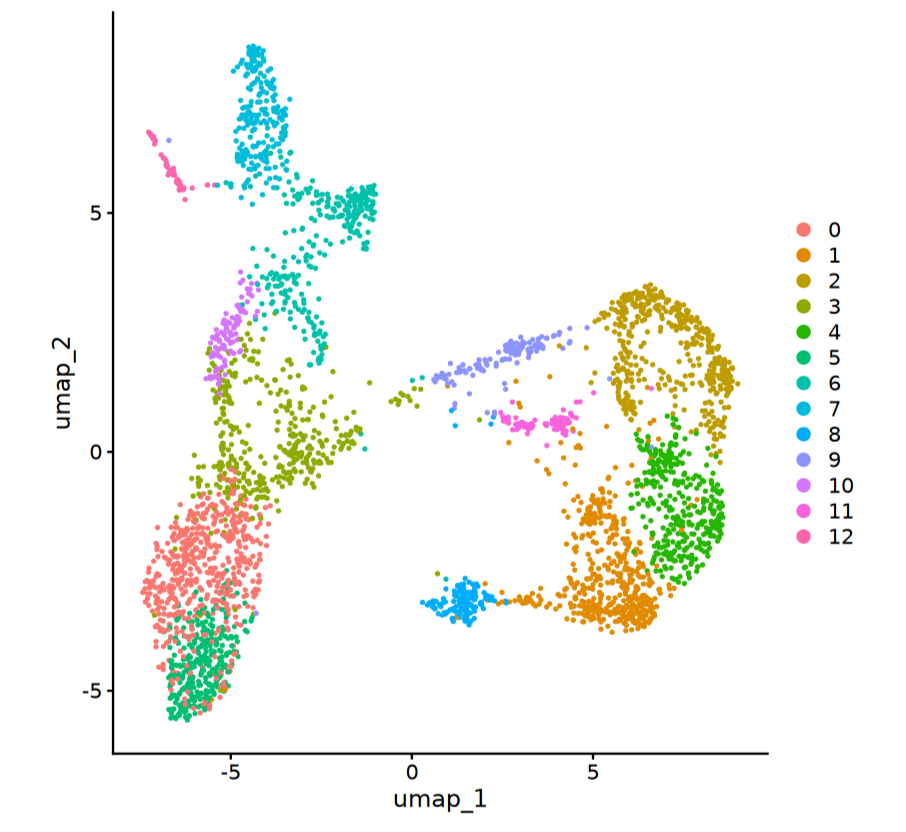
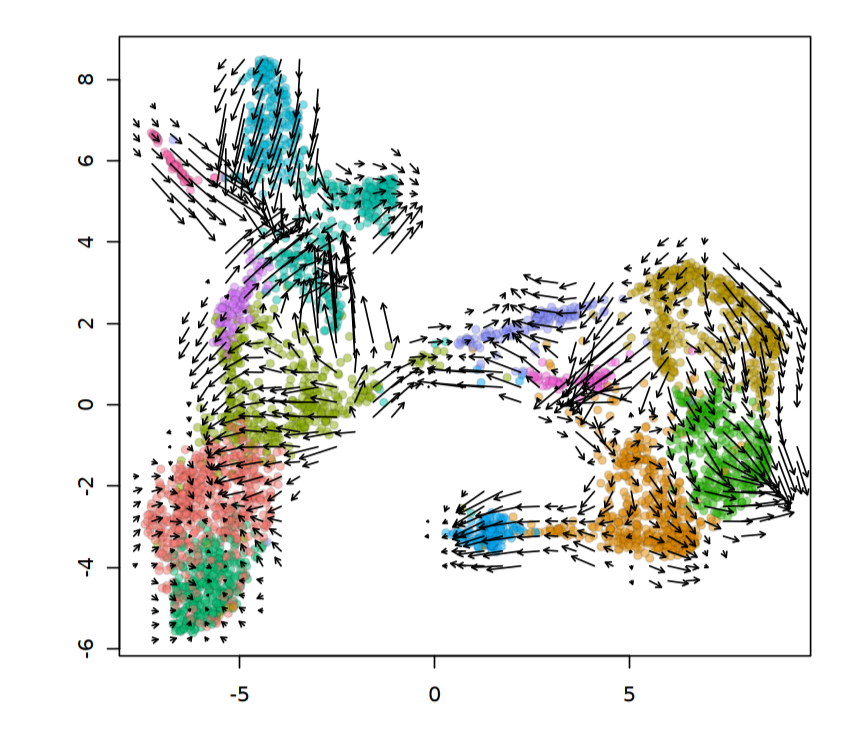
##特定基因速率可视化
gene = "Rab10"
RunVelocity(velo, deltaT=1, kCells=25, fit.quantile=0.02, old.fit=vel,
cell.emb=emb, cell.colors=cell.colors, show.gene=gene, do.par=T)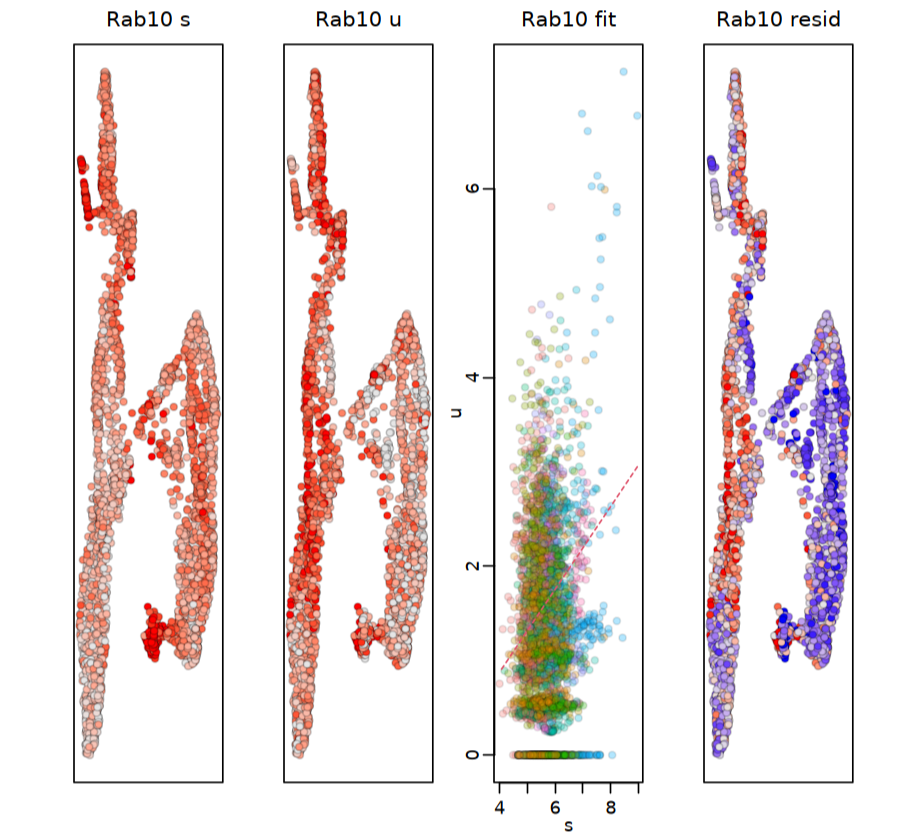

AtomGit 是由开放原子开源基金会联合 CSDN 等生态伙伴共同推出的新一代开源与人工智能协作平台。平台坚持“开放、中立、公益”的理念,把代码托管、模型共享、数据集托管、智能体开发体验和算力服务整合在一起,为开发者提供从开发、训练到部署的一站式体验。
更多推荐

 5
5 0
0- 0



 已为社区贡献1条内容
已为社区贡献1条内容

所有评论(0)