AI驱动药物设计:如何利用Evobind2与AutoDock Vina从头设计高亲和力环肽?
近年来,环肽类分子凭借其兼具小分子药物的优良组织穿透能力和抗体药物的高靶向特异性,正成为创新药研发领域的“新宠”。然而,发现并设计出具有高亲和力的环肽结构,传统上往往需要耗费大量的时间进行试错。
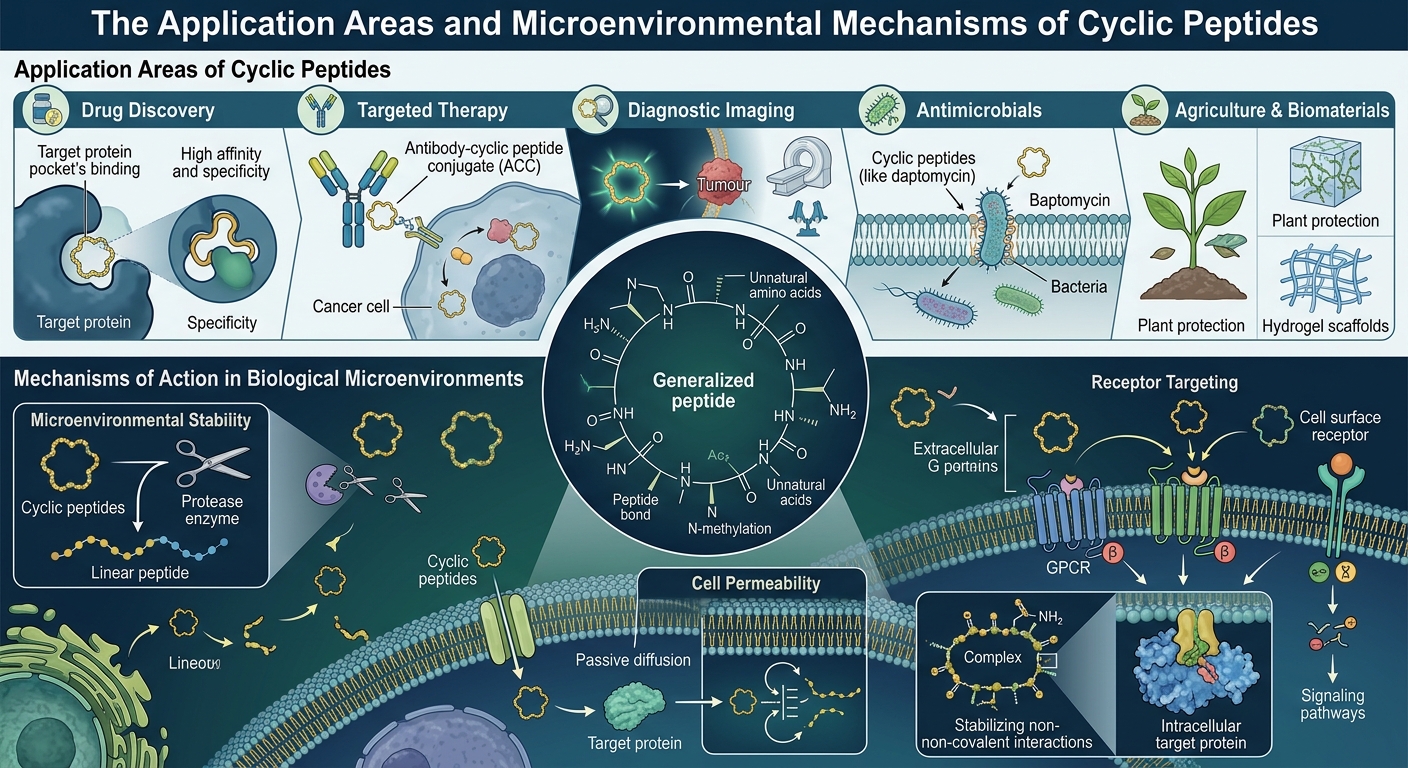
随着AI结构预测与分子对接技术的飞速发展,“计算先行,实验验证”的范式正在重塑这一过程。今天,我们将借助科晶生物的一项从头设计高亲和环肽技术的真实案例,为大家硬核拆解:如何通过计算机辅助与AI大模型,高效、精准地设计并筛选出潜力环肽分子?
一、 明确靶点:精准锁定“攻坚阵地”
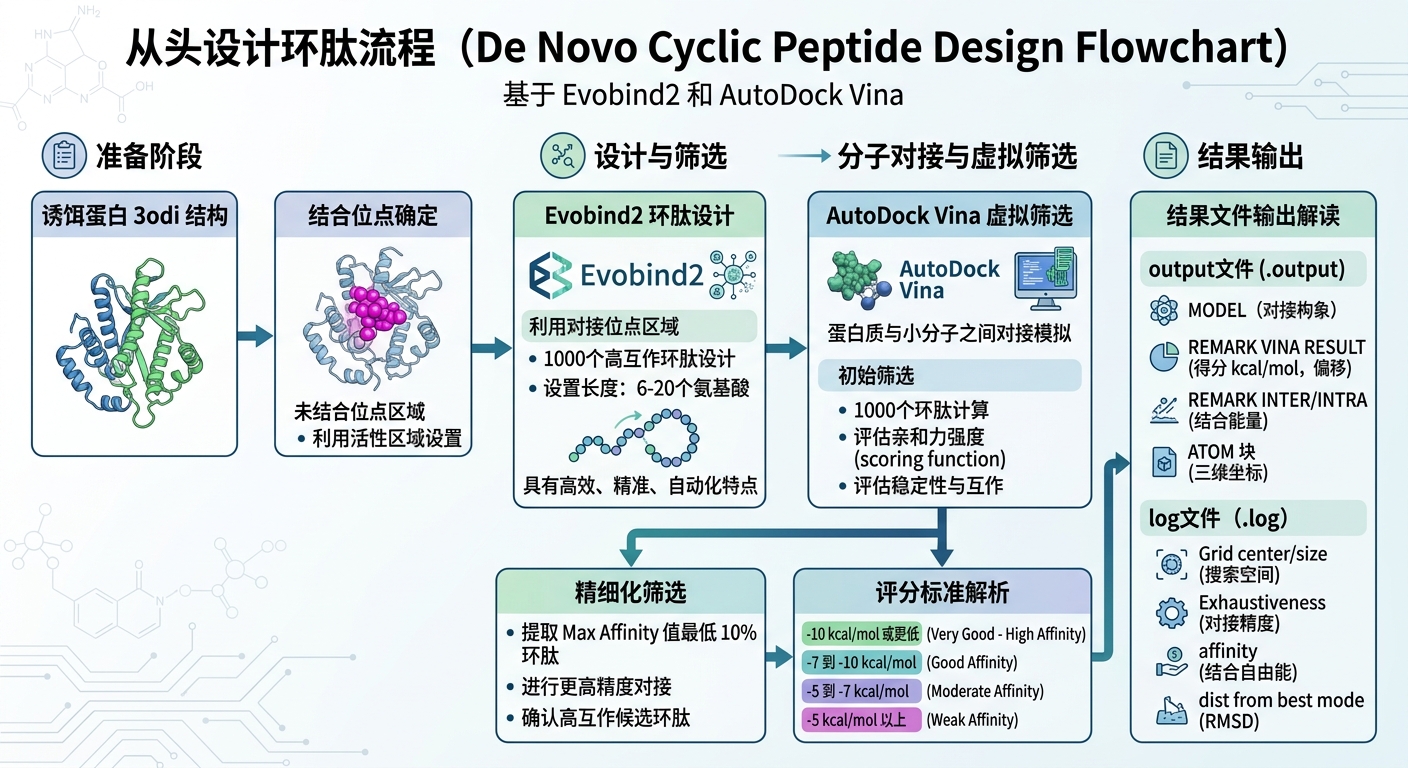
药物设计的核心在于“对症下药”。在科晶生物的这套研发管线中,第一步是结合位点的确定。我们需要利用靶标蛋白与底物结合的活性区域,合理设置未结合位点的范围,以此作为后续结合物设计的“靶心”。
以诱饵蛋白 3odi 为例,这一步需要通过三维结构分析,精准划定对接位点范围。选对结合口袋,就等于成功了一半。

二、 AI从头设计:Evobind2 的“造物”过程
确定了靶心,接下来就需要制造“弹药”。科晶生物在这个环节引入了前沿的深度学习模型 —— Evobind2。
相比于传统的随机生成,Evobind2 模型凭借其高效、精准、自动化的特点,为环肽设计提供了强大的算力支撑。在实际操作中,研究人员将环肽的长度设定为 6-20个氨基酸,通过Evobind2直接在设定的对接位点区域“从头(De novo)”生成了 1000 个具有潜在结合能力的候选环肽。
这一过程极大地提升了早期分子发现的速度与结构多样性,为后续筛选提供了丰富且高质量的“弹药库”。
三、 虚拟筛选与精细过滤:AutoDock Vina 的严苛考验
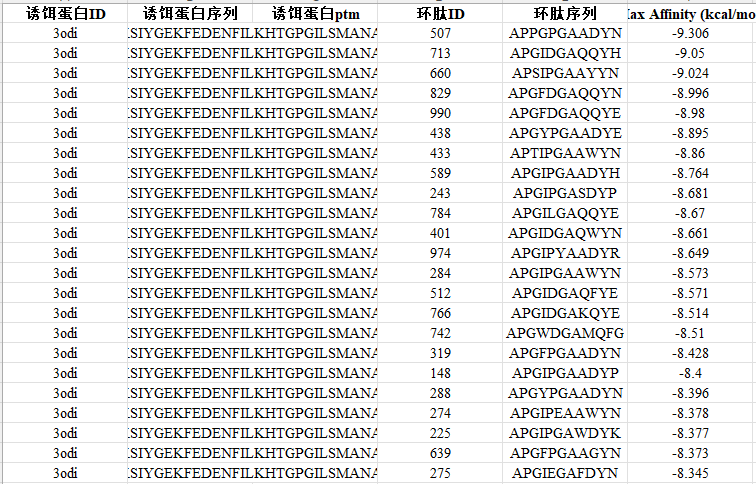
设计出的1000个环肽哪个最好?这就需要用行业金标准软件 AutoDock Vina 来进行分子对接模拟与评分。
AutoDock Vina 采用了优化的经验评分函数(Scoring function),能够综合各种生物物理参数来评估二者的相互作用强度(结合自由能)。科晶生物在这里采用了一种高容错、高精度的“漏斗式”筛选策略:
- 初筛:将1000个候选环肽全部投入对接计算,获取最大亲和力(Max Affinity)值。
- 精筛:提取初筛表现最好的 Top 10% 进入精细化筛选阶段。精筛具有更高的对接精度设定,Max Affinity预测值越低,代表结合越紧密、构象越稳定。
通常,AutoDock Vina的评分被划分为四个梯度:
- >-5 kcal/mol:弱结合(Weak affinity)
- -5 ~ -7 kcal/mol:中等结合(Moderate affinity)
- -7 ~ -10 kcal/mol:良好结合(Good affinity)
- ≤ -10 kcal/mol:非常好的结合(High affinity binding)
严格的筛选不仅能够排查掉无效结构,也能为后续的湿实验(Wet Lab)合成大大节约成本。
四、 硬核拆解:如何看懂对接输出文件?(干货收藏)
对于很多正在进行药物计算设计的研发人员来说,拿到结果文件不知道怎么深入解读是个痛点。在此,我们以该项目的输出结果为例,为大家梳理核心参数。
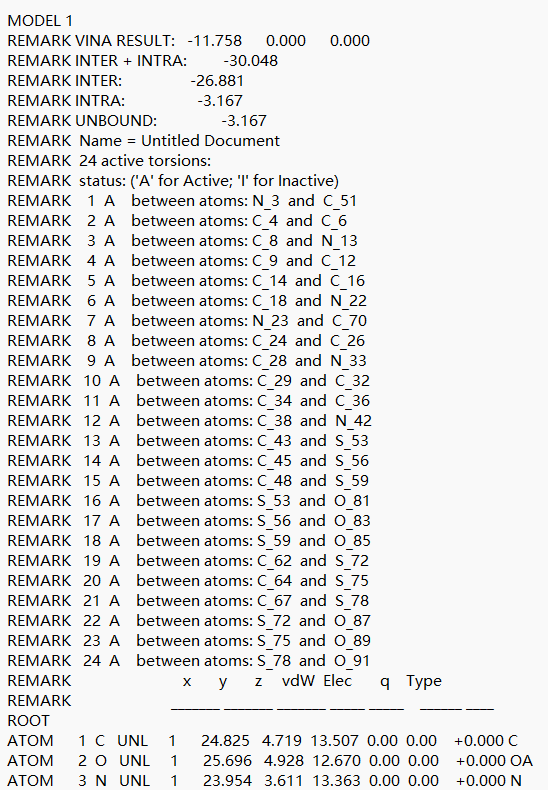
1. .output 文件核心解读:
- VINA RESULT:这是最重要的评分指标。例如结果中显示
-11.758,意味着结合能极低,达到了High affinity极佳结合的水平。后两项数值则代表配体相对于参考位置的平移和旋转偏移。 - REMARK INTER + INTRA:总结合能量(如-30.048),负值越大结合越稳定。它是由分子间能量(配体与受体间直接结合力,如-26.881)与分子内能量(配体自身内部应变能,如-3.167)组成的。
- TORSDOF:代表分子中可旋转键的数量(如文中展示为3个Active torsions),这决定了分子构象的柔性和进入口袋的姿态。
2. .log 文件核心解读:
- Grid center & Grid size:精准定义了对接搜索的“能量盒子”中心坐标(如0.793, -1.066, 0.392)及三维空间尺寸。
- Exhaustiveness:即对接精度。通常参数设为8就能满足基本需求,本案例中将其拉高至 128,这也意味着付出了极大的算力成本,换来了更加精确可靠的最优构象搜索结果。
- RMSD(均方根偏差):用于衡量其他结合模式与最优模式1的偏离距离。通常RMSD < 2 Å被认为是高度相似且合理的对接构象。
五、 结语
从“靶点确证”到“Evobind2从头设计”,再到“高精度Vina虚拟筛选”,这条AI驱动的环肽设计管线,向我们展示了现代计算生物学在药物发现初期爆发出的强大能量。
像科晶生物这样提供系统化、标准化高亲和力环肽设计技术服务的团队,正在帮助更多的科研工作者和医药企业跨越“干湿实验”的鸿沟。将繁重、随机的早期试错交给AI和算力,把宝贵的资金和时间投入到最具有成药潜力的分子验证上,这也正是未来医药创新的大势所趋。
参考文献:
[1] Li Q, Vlachos E.N., Bryant P. Design of linear and cyclic peptide binders of different lengths from protein sequence information. bioRxiv. 2024. p. 2024.06.20.599739.
[2] Trott, O., & Olson, A. J. (2010). AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. Journal of Computational Chemistry, 31(2), 455-461.
[3] Seeliger, D., & de Groot, B. L. (2010). Ligand docking and binding site analysis with PyMOL and AutoDock/Vina. Journal of Computer-Aided Molecular Design, 24(5), 417-422.
[4] O'Boyle, N. M., Banck, M., James, C. A., Morley, C., Vandermeersch, T., & Hutchison, G. R. (2011). Open Babel: An open chemical toolbox. Journal of Cheminformatics, 3(1), 33.

AtomGit 是由开放原子开源基金会联合 CSDN 等生态伙伴共同推出的新一代开源与人工智能协作平台。平台坚持“开放、中立、公益”的理念,把代码托管、模型共享、数据集托管、智能体开发体验和算力服务整合在一起,为开发者提供从开发、训练到部署的一站式体验。
更多推荐



 8
8 0
0- 0



 已为社区贡献28条内容
已为社区贡献28条内容

所有评论(0)