告别盲盒式筛选:AI与扩散模型驱动下的靶标化合物“从头设计”全流程解析
随着2024年诺贝尔化学奖颁发给David Baker的蛋白质从头设计(De novo protein design)研究,计算生物学与AI正式迎来了属于自己的黄金时代。结构生物学不再局限于对天然蛋白的静态解析,而是正在向“动态生成”与“精准定制”跃迁。
然而在硬币的另一面,当我们拥有了具有特定功能的靶标蛋白后,如何快速找到能够与之产生高亲和力结合的化合物分子,依然是摆在无数科研工作者和药企面前的一道难题。传统的做法是依赖庞大的化合物库进行高通量虚拟筛选,但这无异于“大海捞针”,且极大地受限于现有库中分子的骨架多样性。
那么,有没有一种方法,可以跳出现有化合物库的限制,直接针对我们感兴趣的靶点口袋,“量身定制”出自然界中或许还不存在,但亲和力极高的新型化合物呢?
近期,科晶生物交出了一份极具实战参考价值的答卷——基于扩散模型(Diffusion Models)与深度学习的化合物从头设计(De novo compound design)技术方案。今天,我们就来深度拆解这一套兼具高效与高准确率的技术Pipeline。
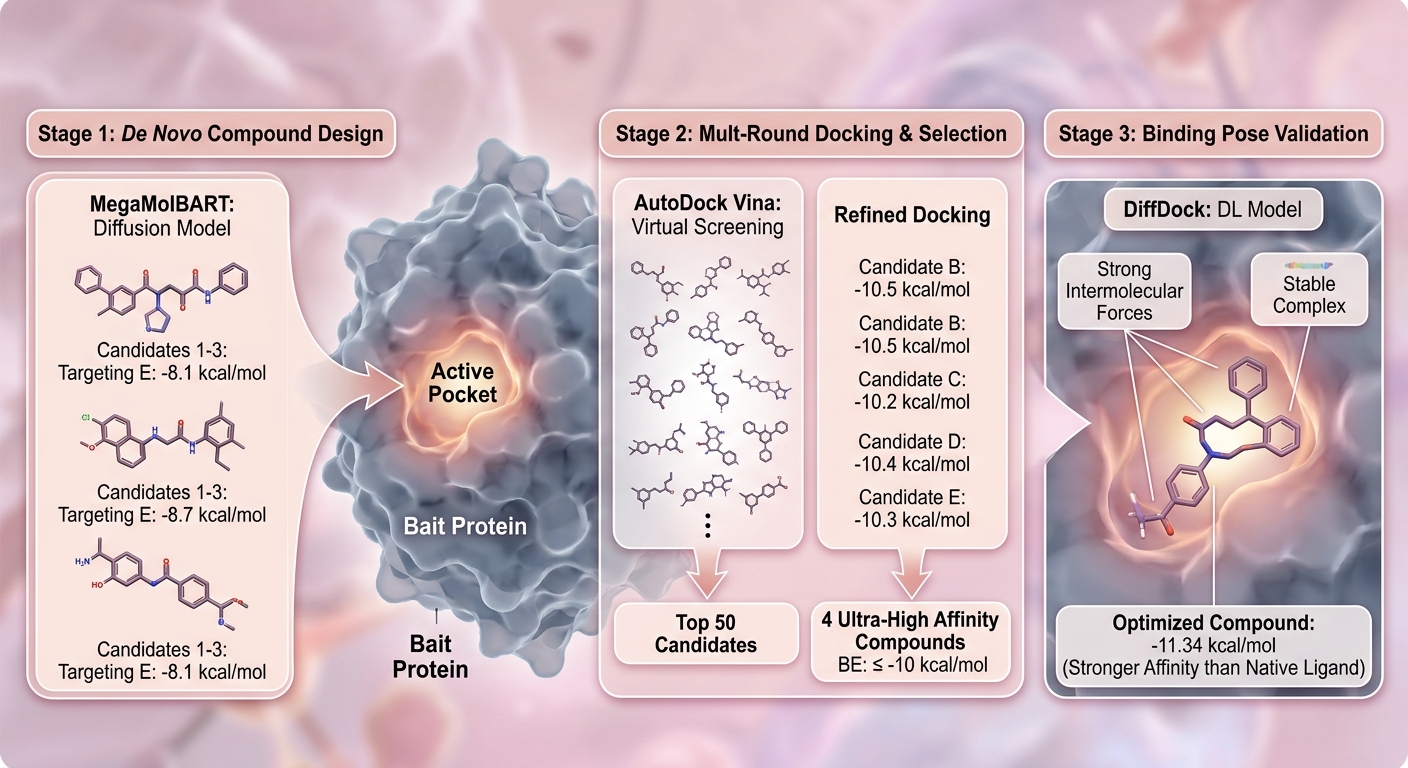
化合物从头设计流程思路
第一步:精准锁定“靶心”——结合口袋的确立
一切精准设计的反击,都始于靶心的精确定位。针对诱饵蛋白的结合口袋,科晶生物的方案采用了双轨制策略:
- 情境一(已知信息丰富):如果研究者有已知的天然配体信息,系统会直接利用诱饵蛋白与原生配体的复合物结构来精准确定结合口袋。这是最准确、最理想的启航点。
- 情境二(全新靶点探索):面对缺乏配体信息的未知或全新靶点,系统会利用算法预测潜在的结合口袋,为无先验信息的靶点打开突破口。
第二步:从破局到生成——应用 MegaMolBART 进行分子“凭空捏造”
找到了口袋,接下来就是“造物”环节。这里引入了核心的AI引擎——MegaMolBART。
在科晶生物的技术流程中,MegaMolBART 是一款基于等变扩散模型(Equivariant Diffusion Model)的结构驱动药物设计工具。它直接打破了传统化合物库的桎梏,支持在指定位置从头生成、修复或优化分子结构。
对于每一个蛋白靶点,算法会定向生成 300 个全新的候选化合物。这些化合物不仅在空间结构上契合口袋,还会通过内置的进化算法提升其药物可及性(Druggability)与化学合成的难易度(Synthesizability)。随后,生成的分子会经过严谨的能量最小化、加氢及电荷分配处理,转化为下游对接所需的三维构象。

第三步:双重过滤网——AutoDock Vina 高通量与精细化对接
AI生成的分子不仅要“长得像”,更要“贴得紧”。在这个环节,物理学原理的评分函数回归主场。
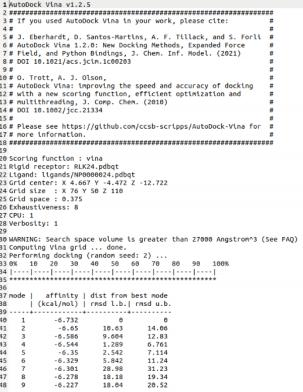
- 初筛(高通量分子对接):首先使用 AutoDock Vina 进行首轮大规模筛选,快速剔除无效构象。系统会提取亲和力极值(Max Affinity)排名前 50 的化合物,进入下一轮。
- 精筛(精细化分子对接):进入 Top 50 的候选者,将接受精度最高、消耗计算资源更大的精细化对接。Vina 的评分依据严谨的物理热力学原理(包含分子间与分子内相互作用能),结合能越负表示结合越稳定,RMSD值确保结构的可靠性。
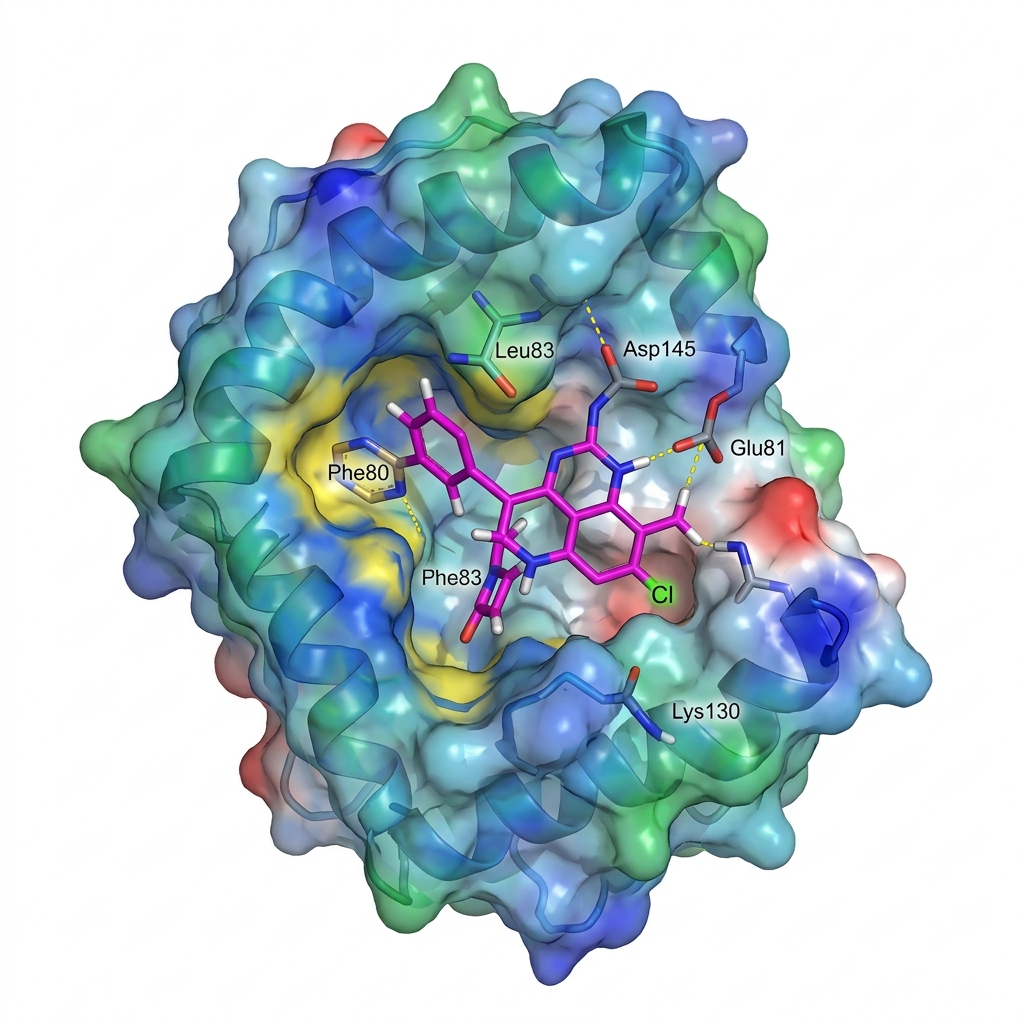
第四步:深度学习终极交叉验证——DiffDock 预测结合构象
为了进一步降低假阳性率,科晶生物的体系还创新性地引入了 DiffDock 作为最终把关。
DiffDock 是麻省理工学院(MIT)团队研发的基于深度学习和生成扩散模型的蛋白-配体对接先锋算法。传统分子对接容易陷入庞大构象空间的算力瓶颈,而 DiffDock 巧妙地通过逐步优化配体的平移、旋转和扭转角度,直接生成符合物理约束的最优姿势。它在处理未知靶点时展现出了强大的泛化能力,不仅节省了算力资源,更为前序筛选的结果上了一道坚实的“双保险”。
实战数据表现:“从头设计”效能如何?
谈及技术服务,终究要回到实战数据上。根据科晶生物最新一期从头设计化合物的结题报告数据:
在 AutoDock Vina 的评分体系中(-10 kcal/mol 以下为非常好的结合,-7 到 -10 为良好结合),该 Pipeline 针对特定靶点经过高通量筛选后,发现了 4 个评分在 -10 kcal/mol 或更低 的超高亲和力分子。而在随后极高精度的 Top 50 精细化复筛中,这 4 个强相互作用分子依然稳如泰山,其中最高结合能甚至达到了惊人的 -11.34 kcal/mol。
这一数据充分证明了结合了MegaMolBART与双轨制对接(Vina+DiffDock)的Pipeline,有极高的概率在实验室湿实验中获得出色的互作验证(Hit rate)。
结语
从盲搜验证到“靶向定做”,计算生物技术正在以肉眼可见的速度重塑科研的边界。科晶生物推出的这套从头设计化合物技术服务,通过前沿扩散模型与经典物理引擎的有机串联,极大地缩短了早期先导化合物发现的周期。
对于正在寻找破局点、或是研究“难成药靶点(Undruggable targets)”的科研人员来说,拥抱这类成熟且经过验证的AI驱动型定做服务,或许是将手中靶点转化为实际科研成果的一条高效捷径。
参考文献:
1.Watson, J. L., et al. (2023). De novo design of protein structure and function with RFdiffusion. Nature, 620, 1089-1099.
2.Haley, O. C., Harding, S., Sen, T. Z., Woodhouse, M. R., Kim, H.-S., Andorf, C. (2024). Application of RFdiffusion to predict interspecies protein-protein interactions between fungal pathogens and cereal crops. bioRxiv.
3.Watson, J. L., Juergens, D., Bennett, N. R., Trippe, B. L., Yim, J., Eisenach, H. E., ... & Baker, D. (2023). De novo design of protein structure and function with RFdiffusion. Nature, 620(7685), 1089-1098.
4.Baek, M., DiMaio, F., Anishchenko, I., Dauparas, J., Ovchinnikov, S., Lee, G. R., ... & Jumper, J. (2021). Accurate prediction of protein structures and interactions using a three-track neural network. Science, 373(6557), 871-876.
5.Lin, Z., Rigden, D. J., & McGuffin, L. J. (2023). Language models of protein sequences at the scale of evolution enable accurate structure prediction. Science, 379(6631), 1123-1130.
6.Dauparas, J., Anishchenko, I., Bennett, N. R., Bai, H., Ragotte, R. J., Milles, L. F., ... & Baker, D. (2022). Robust deep learning-based protein sequence design using ProteinMPNN. Science, 378(6615), 49-56.
7.Yan Y , Tao H , He J ,et al.The HDOCK server for integrated protein-protein docking[J].Nature Protocols, 2020, 15(Suppl 25):1-24.
8.Abramson, J., Adler, J., Dunger, J. et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3.Nature.630,493-500(2024).
9.van Kempen M, Kim S, Tumescheit C, Mirdita M, Lee J, Gilchrist CLM, Söding J, and Steinegger M. Fast and accurate protein structure search with Foldseek. Nature Biotechnology, 2023.

AtomGit 是由开放原子开源基金会联合 CSDN 等生态伙伴共同推出的新一代开源与人工智能协作平台。平台坚持“开放、中立、公益”的理念,把代码托管、模型共享、数据集托管、智能体开发体验和算力服务整合在一起,为开发者提供从开发、训练到部署的一站式体验。
更多推荐



 5
5 0
0- 0



 已为社区贡献28条内容
已为社区贡献28条内容

所有评论(0)