告别化学炼丹术!让AI读懂反应、预测产率,到了哪一步?《AI4S实战派》第六期回顾
作为业内首个多学科、系统化的AI4S工程实战宝典,“AI4S实战派”栏目立足开放生态、持续演进,致力于“手把手”带你率先跑通科学大模型,将复杂、多学科的AI模型转化为能跑、能用、能创新的生产力工具,帮助科研人员和开发者零门槛上手,加速科学新发现。
化学反应不是单变量事件:底物、试剂、溶剂、温度、时间,5 个维度互相耦合,组合空间巨大到化学家也只能靠直觉。
AI想把这件事规模化,第一步不是预测,而是翻译。把一场化学反应,写成机器能读的句子。
5月7日晚,在《AI4S实战派》第六期暨物质科学专题第一课中,广州国家实验室助理研究员、广东省青年拔尖人才张崇焕以“让AI读懂化学:从表征到反应预测”为主题,沿着AI for化学的发展脉络,一路讲到一份真实反应数据的产率预测代码。既有学科视野,也有硬核动手过程。
今天,我们来一起温故知新(也欢迎在“上海科学智能研究院”视频号收看直播回放):
AI4S把化学家的直觉规模化
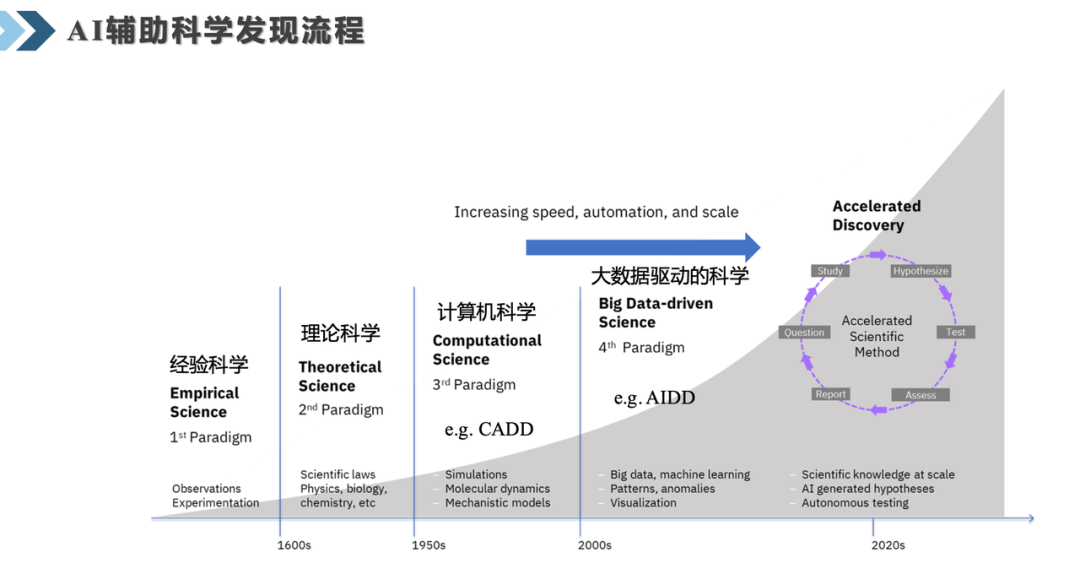
张崇焕在开篇先讲述了科学研究范式经历过的四次跃迁:早期靠观察和实验积累的经验科学,1950年前后兴起的计算机辅助科学,近20年逐渐主导的大数据驱动,再到最近5–10年正在发生的AI加速科学发现。
第四阶段的关键变化在于:AI不再是工具栏里的某一项,而是被深度嵌入了科研闭环;提出问题、获取知识、设计实验、沉淀成果,每一步都有AI的身影。
化学,恰好是检验这件事最严苛的场景之一。一个反应里底物、试剂、溶剂、温度、时间相互交织,因果关系复杂到手动试错根本跟不上节奏。在科研范式的第四阶段,AI不再是工具,而是科研流程的一部分。AI4S不是要取代化学家的直觉,而是要把直觉规模化。
先翻译再预测:分子和反应的四种表征方法
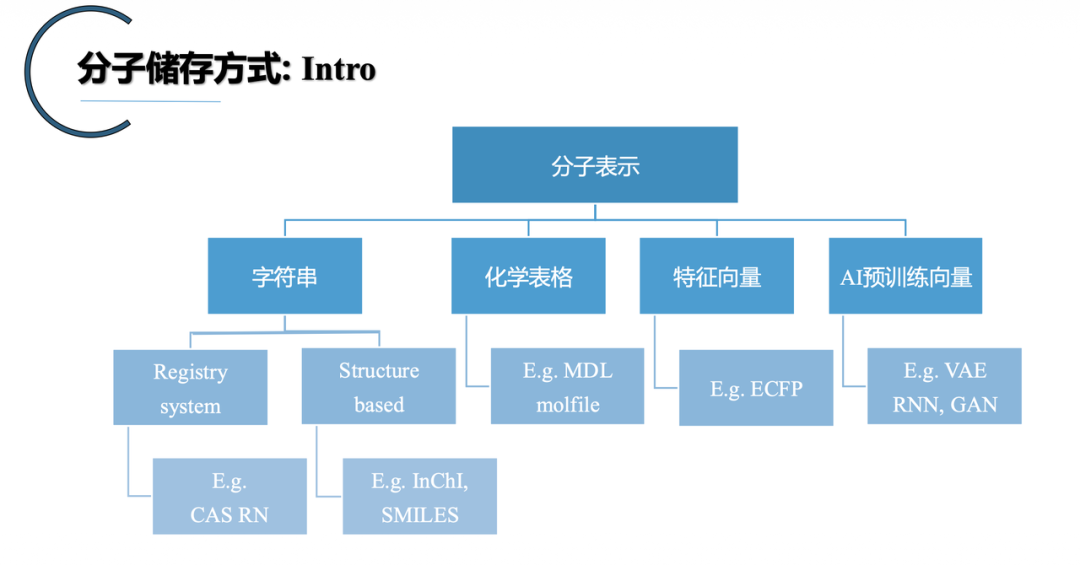
要让AI读懂化学,第一步是把分子和反应翻译成机器能处理的形式。张崇焕梳理了主流的四种表征:
字符串形式:化学式、IUPAC、CAS、SMILES、InChI。其中SMILES是当下最主流的应用,配合canonical SMILES 还能保证每个分子有唯一表达。
表格形式:以molfile为代表,可以记录三维坐标,能在计算化学软件里双向还原结构。
分子指纹:Morgan Fingerprint是经典代表,把分子压缩成0/1二进制向量,可以做相似度分析、推导合成条件、堆叠其他描述符。
图或网络形式:用顶点和边描述分子,是图神经网络的天然输入。
到了反应层面,主要是Reaction SMILES(用>区分底物和产物)和反应指纹(拼接或按位相加分子指纹)。后者按位相加这种看起来粗暴的操作,反而能让计算机运算更顺畅、保留更多信息。
数据来源上,USPTO、Patstat、Reaxys、SciFinder 是AI for化学的“训练粮仓”,然而全都存在脏数据。因此清洗是这个领域的硬功夫之一。
化学反应预测的三层难度:第三层还是无人区
如果说表征是“输入语法”,那预测就是“输出能力”。张崇焕把化学反应预测任务分成了三个层级,难度递增:
Deployment(部署级):可以直接用现有数据库训练、立即落地的任务,比如逆合成、产物预测、反应分类。这一层模型已经成熟,工业界大量在用。
Development(发展级):已有数据不够,需要通过高通量实验或常量化学积累更多数据,比如反应条件预测和优化。
Discovery(发现级):反应机理发现、设计全新反应。现有数据库几乎给不出帮助,需要从文献获取少量先验,再大规模做实验补数据。
第三层,是AI for Chemistry当下的核心难题。

AI能替生化环材人干什么?
抽象的“三层难度”听起来还是有点远。具体到生化环材的实验台上,AI现在能接手两件最核心的“苦力活”。
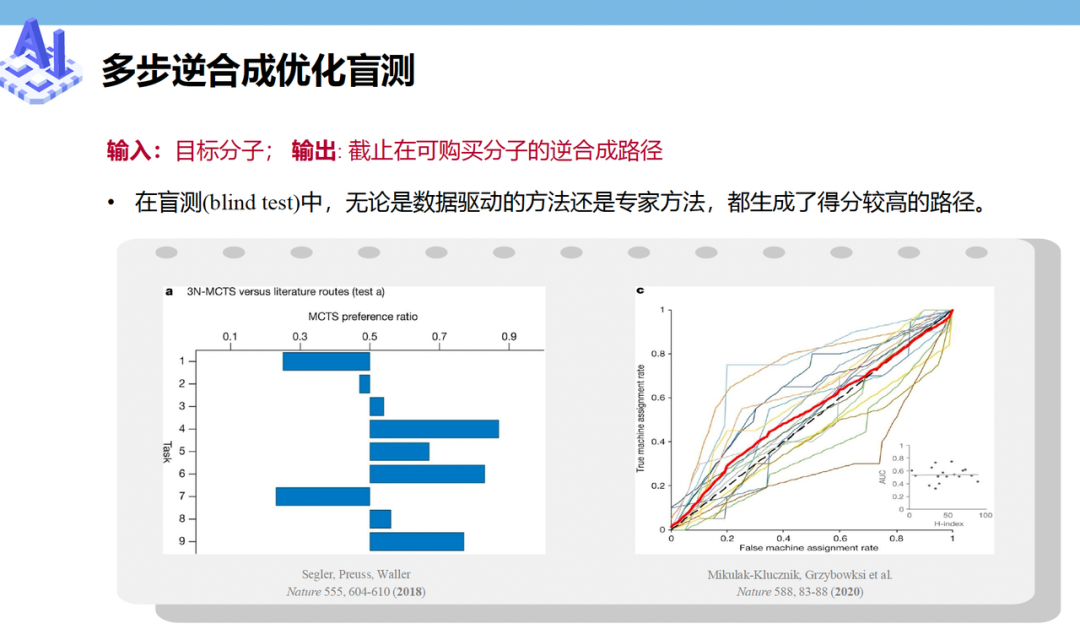
其中一件,是从“找药”走向“做药”的逆合成预测(Retrosynthesis)。
已知一个目标产物,比如一种新设计的抗癌药,AI能像剥洋葱一样,反推它需要哪些基础底物、经过几步反应才能合成出来。听起来有点玄乎,但成绩单其实已经很硬:目前 AI 设计的逆合成路线,在图灵测试中已经能够达到与人类化学家相近的水平。很多时候,化学家看着AI给出的合成方案,已经分不清这是同行做的,还是模型生成的。
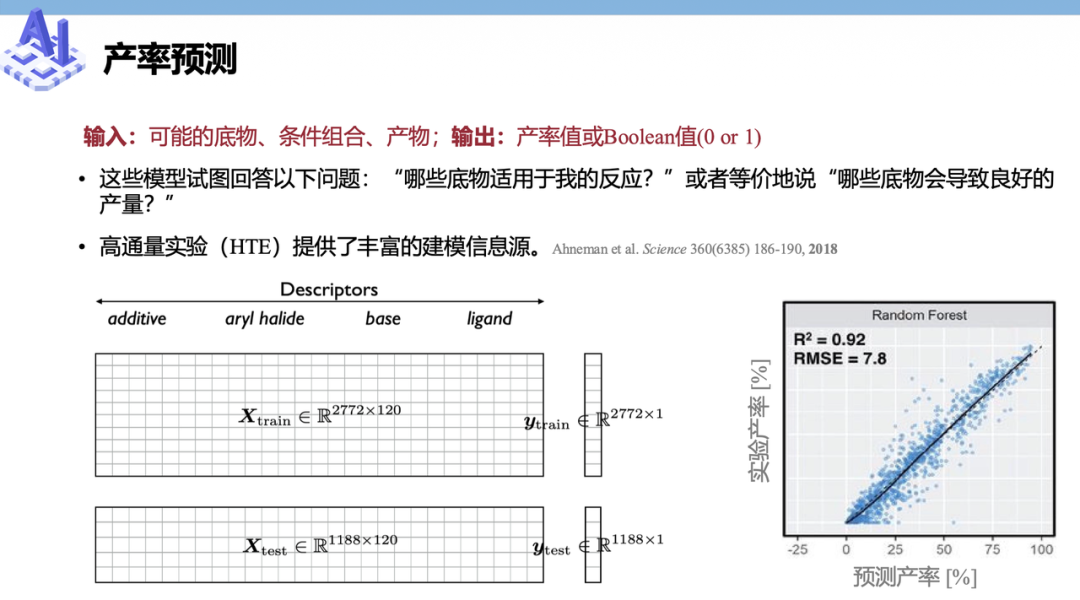
另一件,则是能够省下试剂成本、也省下“肝实验”时间的产率预测(Yield Prediction)。
这是一个与发论文直接挂钩的任务。给定一组底物和一系列候选条件,包括催化剂、溶剂、温度等,AI可以直接给出每种组合的预测产率。研究者不再需要盲目遍历几十种、甚至上百种实验条件,而是只需要优先验证AI跑分最高的Top 5方案,就能大幅提高实验效率、降低试错成本。
原文95%,直播跑出57%,中间差的是工程力
理论之后,张崇焕切到Notebook,在线跑通了一份完整的产率预测案例。
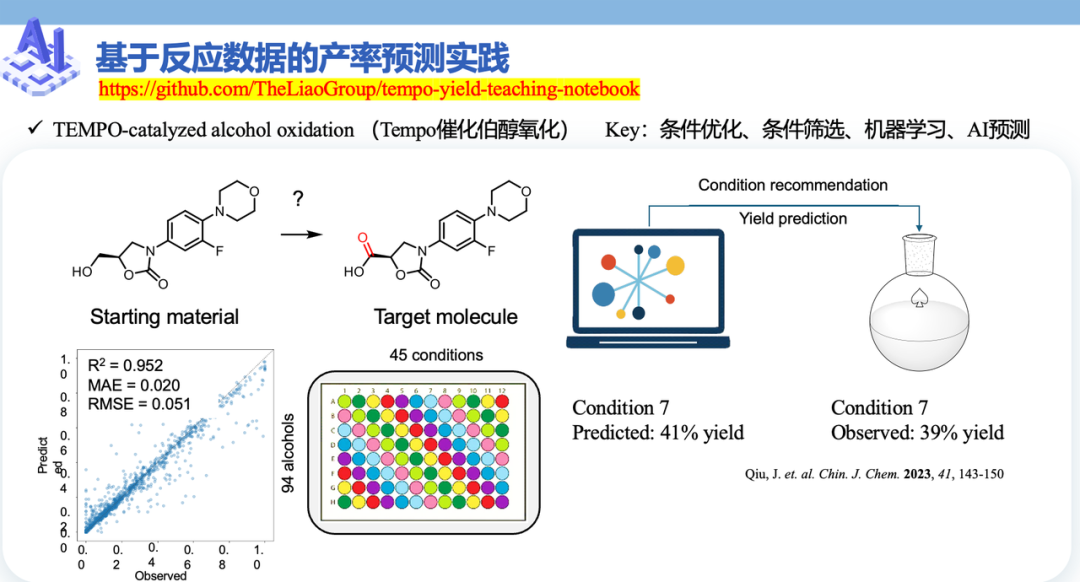
案例选的是2023年发表的Tampo氧化剂催化的伯醇氧化反应(Qiu, J. et. al. Chin. J. Chem. 2023, 41, 143-150)。这一任务属于典型的“单底物生成单产物”问题,配套提供了45组不同反应条件的实验数据,原文测试集的R²高达95%。
完整流程是:先用pandas读取底物、条件和反应信息三张表,再将SMILES转为canonical SMILES,并通过RDKit生成Morgan Fingerprint;随后,把底物与条件的指纹按位相加构成反应指纹,必要时再加入DFT描述符,最终拼接成反应级特征矩阵,并使用Random Forest Regressor建模。
最终跑出来的测试集R²是57%,与论文中的95%还有不小差距。
张崇焕没回避这件事,而是把它讲透:特征工程、数据量、模型选择,每一步都还有提升空间,这才是落地的真实姿态。 模型能跑通只是入口,把效果调到95%,中间差的那38个百分点,才是真正的工程能力。
而当模型可用之后,它的价值也会非常直接:面对一个新的底物,系统可以自动枚举多组候选反应条件,对产率进行预测和排序,再把最高产率对应的方案作为最佳实验条件推荐给化学家。AI改变的,是把过去“赌哪个条件可能成功”,变成了“按概率排序选择最优解”。
完整代码与Notebook已开源至魔搭社区和GitHub上,感兴趣的同学可自行下载复现。
🔗 魔搭:https://modelscope.cn/models/chonghuanzhang/tempo-yield-teaching-notebook
🔗 GitHub:https://github.com/TheLiaoGroup/tempo-yield-teaching-notebook
几条判断和建议
直播尾声,张崇焕一口气回答了十多个问题,其中有几条判断和建议尤其值得记下来。
关于负数据缺失,现有反应数据库几乎只记录“成功案例”,很少保留失败实验,这会导致模型天然带有偏向性。尤其在产率预测任务里,重要的突破口,反而是主动做实验、系统性收集失败数据。
关于逆合成成功率,AI设计的路线一次性合成成功率确实还不算高,但这类任务本身就不能只看“最终是否一步到位”。相比之下,单步反应的TOP-10准确率,其实更适合作为衡量指标。很多时候,不只是模型需要升级,评价标准本身也需要调整。
关于跨学科转型,生化环材背景的学生,最需要补上的能力,包括Python、机器学习底层原理,以及vibe coding。用大模型辅助写代码,已变成新一代科研工作的基本功。
关于特征选择,他也给出了一个非常实用的经验:数据量大的时候,尽量让模型自己学习特征;数据量小的时候,再结合feature importance和化学先验知识手动“剔噪”。规则其实不复杂,但很多人恰恰把顺序做反了。
🚀 下期预告:从化学反应进入原子尺度模拟
第一课打开了化学反应的入口,第二课要往更微观的尺度走。
华东师范大学化学与分子工程学院教授、紫江优秀青年学者朱通将带来物质科学专题的第二讲。
主题为“用机器学习势函数做原子尺度模拟”,内容将聚焦机器学习势函数的拟合逻辑与精度、旋转不变性与等变性如何嵌入模型,以及主动学习如何用最少标注换来最多信息。
朱通长期推动AI与量子化学理论融合,致力于在原子尺度、亚飞秒分辨率下实现复杂反应机理的高精度动力学模拟。这场直播也会进一步回答一个关键问题:AI究竟是如何把原本耗时数周的分子模拟,压缩到分钟级,同时尽可能不损失精度。

AtomGit 是由开放原子开源基金会联合 CSDN 等生态伙伴共同推出的新一代开源与人工智能协作平台。平台坚持“开放、中立、公益”的理念,把代码托管、模型共享、数据集托管、智能体开发体验和算力服务整合在一起,为开发者提供从开发、训练到部署的一站式体验。
更多推荐
 7
7 0
0- 0



 已为社区贡献4条内容
已为社区贡献4条内容

所有评论(0)