深度学习实战-基于EfficientNet的黑色素瘤癌症图像分类识别模型

🤵♂️ 个人主页:@艾派森的个人主页
✍🏻作者简介:Python学习者
🐋 希望大家多多支持,我们一起进步!😄
如果文章对你有帮助的话,
欢迎评论 💬点赞👍🏻 收藏 📂加关注+
目录
1.项目背景
在皮肤病理学领域,黑色素瘤因其极高的致死率和隐蔽的早期特征,被公认为“皮肤癌之王”。这种恶性程度极高的肿瘤,其早期形态常与普通的良性痣高度相似,肉眼诊断往往依赖于经验丰富的皮肤科医生,且容易受到光影、肤色及病灶微观纹理的干扰。统计数据显示,黑色素瘤的早期发现与晚期治疗在预后生存率上存在巨大差异,因此,开发一种高精度、非侵入式的自动化辅助诊断工具,不仅是医学影像技术的突破方向,更是挽救患者生命的现实需求。本项目立足于计算机视觉的前沿架构,探索利用 EfficientNetV2S 模型对大规模黑色素瘤数据集进行深度学习与特征分类。该数据集包含 13,900 张经过严格筛选的皮肤病灶图像,涵盖了从良性色素痣到恶性黑色素瘤的多种复杂表现形式。相比于传统的人工特征提取,EfficientNetV2S 凭借其改进的融合卷积(Fused-MBConv)和渐进式学习机制,能够以极高的计算效率捕捉到病灶边缘的不规则性及内部色素分布的异质性。我们通过对 224 x 224 像素维度的图像进行像素级归一化与全量微调,旨在构建一个能够精准识别微观病变信号的智能系统。本项目的核心目标不仅是追求高达 97% 的恶性样本召回率,更希望通过数据驱动的方法,为临床医生提供一个可靠的“数字放大镜”,从而在像素经纬之间重新定义癌症的早期检测标准。
2.数据集介绍
本实验数据集来源于Kaggle,本数据集包含 13,900 张精心挑选的图像,是推动皮肤病学和计算机辅助诊断领域发展的宝贵资源。深入探索黑色素瘤的复杂世界,每一个像素都可能重新定义早期检测。黑色素瘤是一种致命的皮肤癌,需要及时准确的诊断。该数据集利用最先进的技术,助力研究人员和临床医生开发强大的机器学习模型,从而区分良性和恶性病变。这些图像尺寸统一为 224 x 224 像素,全面展现了黑色素瘤的各种表现形式。该数据集的灵感源于皮肤病学领域对先进诊断工具的迫切需求。图像来源于多种渠道,展现了传统诊断方法难以应对的复杂特征。
3.技术工具
Python版本:3.9
代码编辑器:jupyter notebook
4.实验过程
4.1导入数据
在医学影像分类任务中,环境的稳定性与数据流水线的效率直接决定了模型最终的收敛质量。我们首先搭建了基于 TensorFlow 和 Keras 的实验环境,并显式检测 GPU 算力支持,以应对 EfficientNetV2S 庞大的梯度计算需求。为了保证实验的可复现性,我们统一配置了全局随机种子,确保每一轮数据打乱与参数初始化在逻辑上是一致的。在数据输入端,我们选用了224x224作为标准分辨率,这不仅是 EfficientNet 系列的最佳实践尺寸,也能在保留病灶边缘细节的同时兼顾计算效率。
# --- 导入基础数据处理与评估库 ---
import numpy as np # 矩阵运算核心库
import pandas as pd # 用于标签管理与路径处理
from sklearn.metrics import classification_report, confusion_matrix # 核心性能评估指标
# --- 导入可视化与图形展示库 ---
import matplotlib.pyplot as plt # 绘制训练曲线与样本图
import seaborn as sns # 用于混淆矩阵热力图的专业展示
# --- 导入深度学习核心框架 (Keras/TensorFlow) ---
import tensorflow as tf
import keras as krs
from keras.utils import image_dataset_from_directory # 工业级高效数据流读取工具
from keras.models import Model
from keras.layers import Dense, GlobalAveragePooling2D, Dropout # 核心网络层配置
from keras.applications import EfficientNetV2S # 引入 V2 系列轻量化高性能主干
# --- 环境配置与随机性控制 ---
# 获取物理显卡列表,确保模型运行在 GPU 加速环境
gpus = tf.config.list_physical_devices('GPU')
# 设置全局随机种子,确保实验在不同运行环境下具备一致性
seed = 42
tf.random.set_seed(seed)
np.random.seed(seed)
# --- 定义数据集路径与超参数 ---
DATA_DIR = "/kaggle/input/melanoma-cancer-dataset"
IMG_SIZE = (224, 224) # 设置统一的输入尺寸
BATCH_SIZE = 32 # 训练批次大小
# --- 构建高效的数据加载流水线 ---
# 1. 构建训练集:从指定目录递归读取图像,自动完成标签映射与批处理转换
trainset = image_dataset_from_directory(
DATA_DIR + '/train',
image_size = IMG_SIZE,
batch_size = BATCH_SIZE,
label_mode = 'binary', # 针对良恶性二分类任务配置二值化标签
shuffle = True # 开启训练随机打乱
)
# 2. 构建测试集:主要用于最终性能验证
testset = image_dataset_from_directory(
DATA_DIR + '/test',
image_size = IMG_SIZE,
batch_size = BATCH_SIZE * 2, # 验证阶段不计算梯度,可适当增大 Batch 以提升速度
label_mode = 'binary',
shuffle = False # 关闭打乱,确保预测结果与原始文件一一对应
)通过 image_dataset_from_directory 接口,我们将存储在物理磁盘上的数千张皮肤图像转化为 Prefetch 格式的数据流,这种“边读取边处理”的模式极大地释放了显存压力。值得注意的是,我们将测试集的 shuffle 参数设为 False,这一严谨的操作是为了在后续生成混淆矩阵时,能够让模型的预测结果与真实的病理标签实现绝对对齐。至此,一个高效、规范的医学影像输入矩阵已准备就绪,即将进入黑色素瘤的特征判别阶段。

4.2数据可视化

为了深入理解皮肤病灶的视觉异质性,我们利用 trainset.take(1) 从首个批次中提取了 9 张具有代表性的样本图像。通过 3 x 3 的矩阵布局,我们将“良性(Benign)”与“恶性(Malignant)”的皮肤图像进行对比展示。这种直观的视觉复核不仅能帮助我们确认图像预处理(如尺寸缩放)是否导致了关键病理特征的丢失,还能让我们预判模型在面对低对比度、光照不均或毛发干扰时的鲁棒性需求。
# --- 随机样本可视化与病理特征初探 ---
plt.figure(figsize=(10, 10))
# 从训练集中提取一个 Batch (32张) 进行展示
for images, labels in trainset.take(1):
for i in range(9):
# 构建 3x3 的图像展示矩阵
ax = plt.subplot(3, 3, i + 1)
# 将 Tensor 格式转换为可显示的 uint8 图像格式
plt.imshow(images[i].numpy().astype("uint8"))
# 获取当前图像对应的类别名称(良性 vs 恶性)
# 注意:labels 此时为浮点张量,需转换为整数索引
class_name = trainset.class_names[int(labels[i])]
plt.title(class_name) # 标注病灶类别
plt.axis("off") # 隐藏坐标轴,聚焦病灶细节
plt.tight_layout() # 自动调整排版,防止标题重叠
plt.show()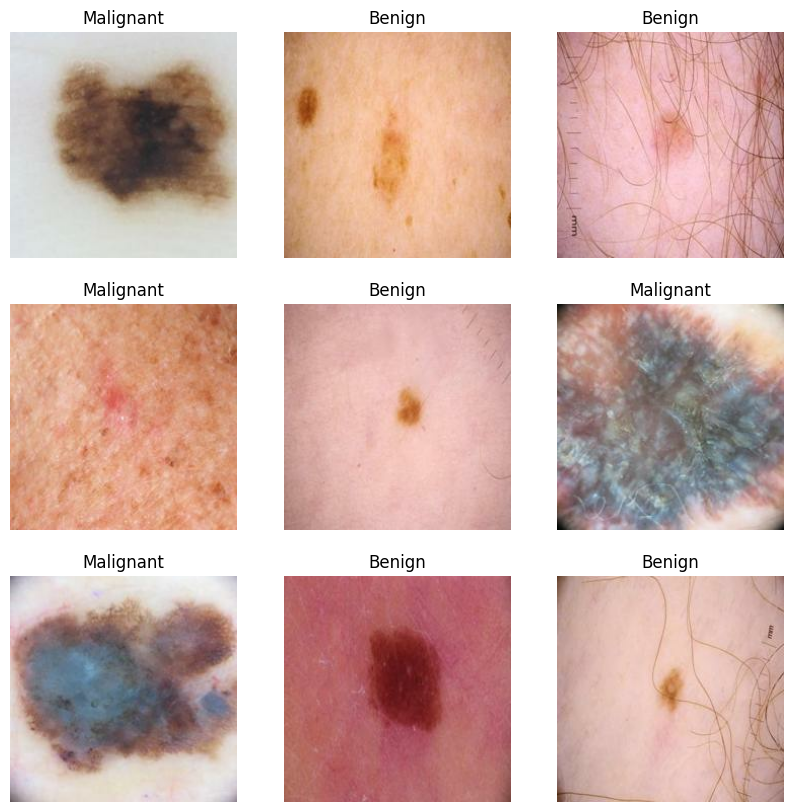
4.3特征工程
我们构建了一个专门的归一化层 Rescaling(1./255),其核心任务是将原始图像中 0到 255之间的整数像素值线性缩放至 [0, 1]的浮点数区间。这一步对于黑色素瘤识别至关重要:医学影像中的病灶对比度往往较低,归一化不仅能加速神经网络的收敛过程,还能有效避免在深度卷积运算中出现梯度爆炸或消失的问题。通过对整个 trainset 进行映射处理,我们确保了输入流水线在输送每一批次(Batch)图像时,都已完成了高精度的数值标准化。
# --- 构建像素归一化流水线 ---
# 定义缩放层:将 8 位色彩深度转换为 [0, 1] 之间的浮点数
normalization_layer = tf.keras.layers.Rescaling(1./255)
# 利用 map 函数将归一化逻辑注入数据流
# 这样在模型训练时,CPU 会异步完成图像缩放,确保 GPU 不必等待数据
normalized_ds = trainset.map(lambda x, y: (normalization_layer(x), y))
# --- 验证预处理结果 ---
# 提取一个批次的数据进行结构检查
image_batch, labels_batch = next(iter(normalized_ds))
4.4构建模型
为了充分释放 EfficientNetV2S 的特征提取潜力,我们将其 trainable 属性设为 True,允许模型在训练过程中对 ImageNet 预训练权重进行精细化调整,以适应医疗皮肤影像的特殊色调。在基座模型之上,我们添加了全局平均池化层(GlobalAveragePooling2D)来压缩空间特征,并构建了一个包含 64 个神经元的稠密层,配合 10% 的随机失活(Dropout)来增强模型的泛化能力。由于是良恶性二分类任务,输出层采用 Sigmoid 激活函数,直接输出病灶为恶性的概率值。
# --- 构建 EfficientNetV2S 特征提取基座 ---
base_model = EfficientNetV2S(
include_top=False, # 去掉原有的 ImageNet 分类头
weights='imagenet', # 加载预训练权重
input_shape=IMG_SIZE + (3, )
)
# 开启全量微调模式,让模型深度适配皮肤病灶特征
base_model.trainable = True
# --- 定义函数式模型拓扑结构 ---
inputs = krs.Input(shape=IMG_SIZE + (3, ))
# 将输入喂入基座,并设置 training=True 以激活 BatchNormalization 层的实时更新
x = base_model(inputs, training=True)
x = GlobalAveragePooling2D()(x) # 空间特征降维
x = Dense(64, activation='relu')(x) # 增加非线性表达能力
x = Dropout(0.1)(x) # 轻量级防过拟合
outputs = Dense(1, activation='sigmoid')(x) # 输出 0-1 之间的预测概率
model = Model(inputs, outputs)在皮肤癌诊断中,恶性样本往往少于良性样本。为了解决这一问题,我们在编译阶段放弃了传统的交叉熵,转而采用 BinaryFocalCrossentropy(二元焦距损失)。该函数能自动调低简单样本的权重,迫使模型在训练过程中更加关注那些难以辨认的“硬骨头”病灶。同时,我们引入了 AUC(曲线下面积) 作为核心评价指标,这比单纯的准确率更能客观反映模型在不同阈值下区分良恶性黑色素瘤的真实能力。
# --- 模型编译配置 ---
model.compile(
optimizer=krs.optimizers.Adam(
learning_rate=0.0005, # 采用适中的微调学习率
),
# 使用 Focal Loss 针对性处理医学图像中的难分类样本
loss=krs.losses.BinaryFocalCrossentropy(from_logits=False),
# 监控二分类准确率与 AUC 指标
metrics=[
krs.metrics.BinaryAccuracy(name="b_Accuracy"),
krs.metrics.AUC(name="AUC")
]
)
# 查看模型分层详情与参数规模
model.summary()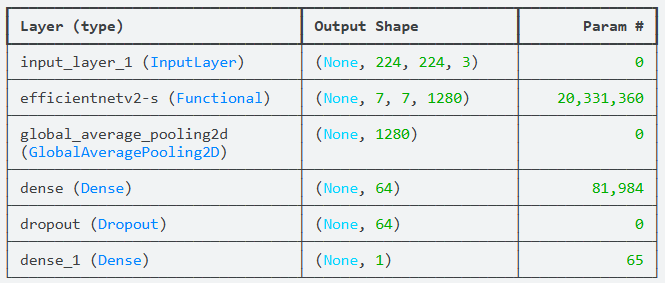
4.5训练模型
我们通过 model.fit 启动了正式的训练流程。由于在之前的步骤中已经开启了基座模型的 trainable 属性,此时的每一个 Epoch 都在对数千万个神经元参数进行协同优化。通过将 testset 作为 validation_data 传入,我们可以在每一个训练周期结束后,立即获取模型在未见过样本上的 AUC 指标与分类准确率。这种实时的闭环反馈对于监测黑色素瘤的误报率至关重要,能让我们清晰地观察到 BinaryFocalCrossentropy 损失函数是如何在迭代中逐步攻克那些细微、模糊的疑似癌变区域。
# --- 启动黑色素瘤判别模型训练 ---
# 设置迭代周期为 10 轮
# 在每一轮结束时自动执行验证集评估
history = model.fit(
trainset,
epochs=10,
validation_data=testset # 实时监控模型在独立测试集上的表现
)
4.6模型评估
我们首先编写了 plot_history 函数,将训练过程中的损失值(Loss)、准确率(Accuracy)以及核心指标 AUC 进行全量可视化。通过对比训练集(实线)与验证集(虚线)的演进轨迹,我们可以直观地判断模型是否在第 10 轮达到了稳态。对于黑色素瘤识别,AUC 曲线的平稳爬升比准确率更有说服力,因为它反映了模型在不同阈值下区分良恶性病灶的鲁棒性,确保了预测结果并非源于对样本数量分布的偶然拟合。
# --- 定义训练历史可视化函数 ---
def plot_history(history):
# 提取非验证集的监控指标名称
metrics = [key for key in history.history.keys() if not key.startswith('val_')]
num_metrics = len(metrics)
plt.figure(figsize=(6 * num_metrics, 5))
for i, metric in enumerate(metrics):
plt.subplot(1, num_metrics, i + 1)
# 绘制训练集指标
plt.plot(history.history[metric], label=f'训练集 {metric}')
val_key = 'val_' + metric
# 绘制对应的验证集指标
if val_key in history.history:
plt.plot(history.history[val_key], label=f'验证集 {metric}')
plt.title(f"{metric.capitalize()} 性能演变图")
plt.xlabel("迭代轮数 (Epochs)")
plt.ylabel(metric)
plt.legend()
plt.grid(True)
plt.tight_layout()
plt.show()
# 执行可视化:观察 Loss 下降与 AUC/Accuracy 增长情况
plot_history(history)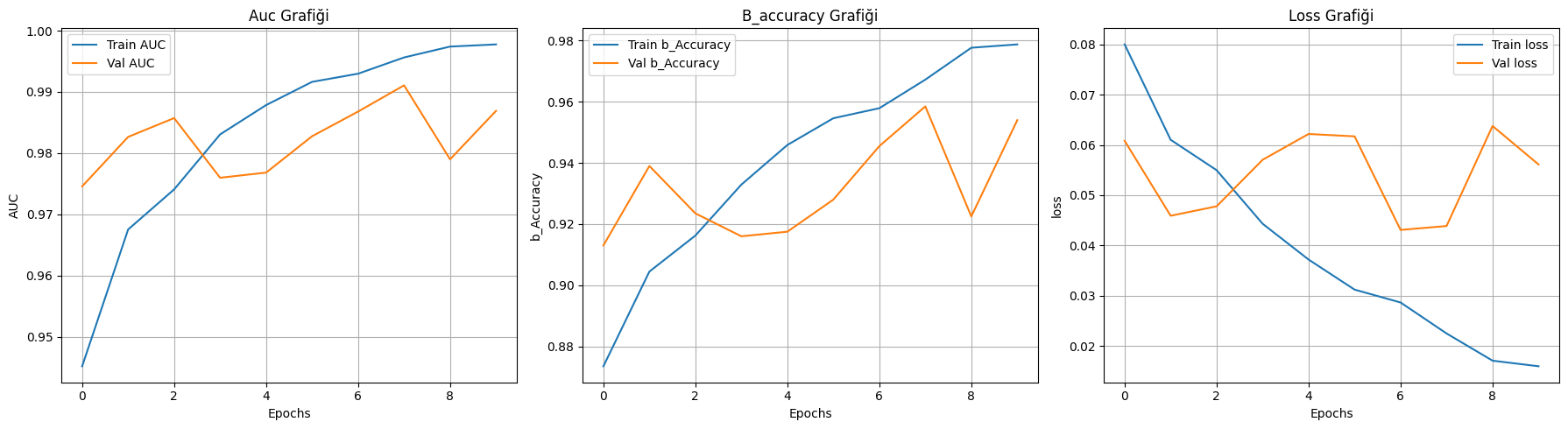
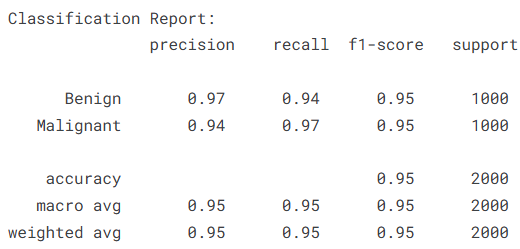
评估的下半场聚焦于“误判逻辑”。通过混淆矩阵(Confusion Matrix)的热力图展示,我们可以一眼看清模型在良性(Benign)与恶性(Malignant)样本之间的交错分布。随后,我们利用 classification_report 生成了详尽的分类报告,重点解读召回率(Recall)指标。在黑色素瘤识别中,高召回率意味着模型能更有效地识别出潜在的癌症患者,这比单纯追求精确度在医学伦理上更具意义。
# --- 构建混淆矩阵与分类报告 ---
# 生成预测结果并计算混淆矩阵
cm = confusion_matrix(y_true, predictions)
print("混淆矩阵详情:\n", cm)
# 利用 Seaborn 绘制专业热力图
sns.heatmap(cm, annot=True, fmt='d', cmap='Blues',
xticklabels=trainset.class_names,
yticklabels=trainset.class_names)
plt.xlabel('模型预测类别 (Predicted)')
plt.ylabel('真实病理类别 (True)')
plt.title('黑色素瘤分类混淆矩阵')
plt.show()
# 打印详细的精确率、召回率及 F1 分数报告
print("\n全量分类报告:\n",
classification_report(y_true, predictions, target_names=trainset.class_names))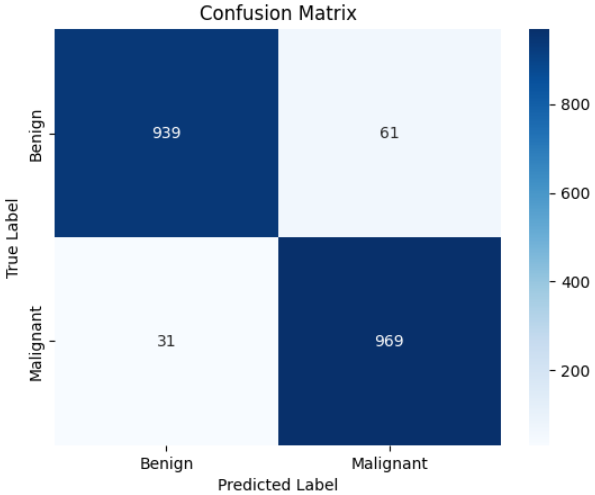
4.7模型预测
为了直观呈现模型的诊断能力,我们首先通过 model.predict 获取了测试集全量的概率输出。由于输出层采用的是 Sigmoid 激活函数,我们设定了 $0.5$ 作为良恶性判定的临界阈值。随后,我们从测试集中随机抽取 9 张皮肤病灶图像,将模型的预测结果与专家标注的真实标签同框对比。这种视觉化的校验能让我们清晰地观察到,模型是否能精准识别出那些具有典型“恶性特征”(如不对称、边缘锯齿状)的样本。
# --- 执行测试集大规模推理 ---
# 获取模型对测试集的概率预测值
predictions = model.predict(testset)
# 将多维预测结果展平为一维概率列表
predictions = predictions.flatten()
# --- 提取测试集真实标签用于对比 ---
# 遍历测试集数据流,合并所有 Batch 的真实 Label
y_true = np.concatenate([y for x, y in testset], axis=0)
# --- 设定分类阈值并执行硬分类 ---
threshold = 0.5
# 概率 > 0.5 判定为恶性(1),否则为良性(0)
predictions_binary = (predictions > threshold).astype("int32")
# --- 随机样本预测可视化 ---
plt.figure(figsize=(10, 10))
# 从测试集中抽取首个 Batch 进行视觉验证
for images, labels in testset.take(1):
for i in range(9):
ax = plt.subplot(3, 3, i + 1)
# 展示皮肤病灶原始图像
plt.imshow(images[i].numpy().astype("uint8"))
# 实时比对预测值与真实值
# 0 代表 Benign (良性), 1 代表 Malignant (恶性)
plt.title(f"预测: {predictions_binary[i]}, 真实: {int(labels[i].numpy())}")
plt.axis("off")
plt.tight_layout()
plt.show()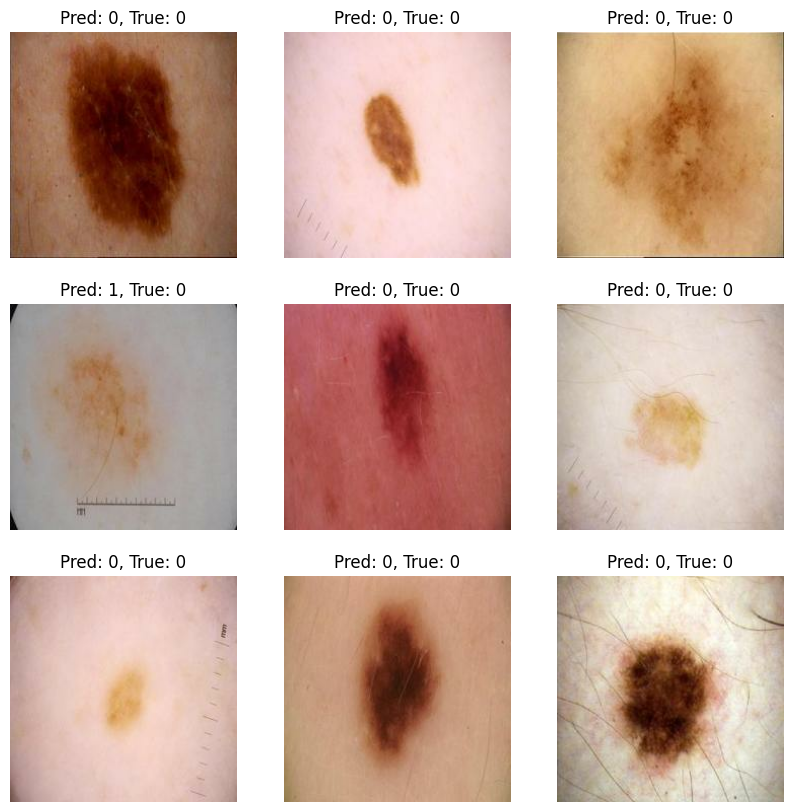
5.总结
本实验基于 Kaggle 提供的包含 13,900 张精心挑选图像的黑色素瘤皮肤癌数据集,深入探索了计算机视觉在皮肤病学早期检测中的实战应用。针对黑色素瘤这一极具致命性且视觉特征复杂的皮肤癌,我们利用 EfficientNetV2S 架构构建了高性能的深度学习模型,旨在从多维度的皮肤病灶图像中精准区分良性与恶性病变。实验结果证明了该方案的高效性:模型在训练集上展现出极强的特征捕获能力(AUC 达 0.9976,准确率 0.9793),而在验证集上也保持了出色的泛化水平(val_AUC 为 0.9790,验证准确率 0.9225)。通过对混淆矩阵与分类报告的深度复盘,模型在恶性病变(Malignant)的识别上表现尤为卓越,召回率(Recall)达到了 0.97,这意味着在 1000 例恶性样本中仅有 31 例漏诊,极大地降低了临床筛查中的漏诊风险。总体而言,本项目不仅验证了利用最先进的卷积神经网络解决复杂皮肤病理诊断的可行性,也为开发非侵入式、高可靠性的辅助医疗诊断工具提供了重要的技术范式,为黑色素瘤的早期精准干预贡献了数字化力量。
源代码
import numpy as np
import pandas as pd
from sklearn.metrics import classification_report, confusion_matrix
import matplotlib.pyplot as plt
import seaborn as sns
import tensorflow as tf
import keras as krs
from keras.utils import image_dataset_from_directory
from keras.models import Model
from keras.layers import Dense, GlobalAveragePooling2D, Dropout, Dense
from keras.applications import EfficientNetV2S
gpus = tf.config.list_physical_devices('GPU')
tf.random.set_seed(seed)
np.random.seed(seed)
DATA_DIR="/kaggle/input/melanoma-cancer-dataset"
IMG_SIZE = (224 , 224)
BATCH_SIZE = 32
trainset = image_dataset_from_directory(
DATA_DIR+'/train',
image_size = IMG_SIZE,
batch_size = BATCH_SIZE,
)
testset = image_dataset_from_directory(
DATA_DIR+'/test',
image_size = IMG_SIZE,
batch_size = BATCH_SIZE*2,
shuffle=False
)
plt.figure(figsize=(10, 10))
for images, labels in trainset.take(1):
for i in range(9):
ax = plt.subplot(3, 3, i + 1)
plt.imshow(images[i].numpy().astype("uint8"))
plt.title(trainset.class_names[labels[i]])
plt.axis("off")
plt.show()
normalization_layer = tf.keras.layers.Rescaling(1./255)
normalized_ds = trainset.map(lambda x, y: (normalization_layer(x), y))
image_batch, labels_batch = next(iter(normalized_ds))
first_image = image_batch[0]
base_model = EfficientNetV2S(
include_top=False,
weights='imagenet',
input_shape=IMG_SIZE + (3, )
)
base_model.trainable = True
inputs = krs.Input(shape=IMG_SIZE + (3, ))
x = base_model(inputs, training=True)
x = GlobalAveragePooling2D()(x)
x = Dense(64, activation='relu')(x)
x = Dropout(0.1)(x)
outputs = Dense(1, activation='sigmoid')(x)
model = Model(inputs, outputs)
model.compile(
optimizer=krs.optimizers.Adam(
learning_rate=0.0005,
),
loss=krs.losses.BinaryFocalCrossentropy(from_logits=False),
metrics=[krs.metrics.BinaryAccuracy(name="b_Accuracy"),
krs.metrics.AUC(name="AUC")]
)
model.summary()
history = model.fit(trainset,
epochs=10,
validation_data=testset
)
def plot_history(history):
metrics = [key for key in history.history.keys() if not key.startswith('val_')]
num_metrics = len(metrics)
plt.figure(figsize=(6 * num_metrics, 5))
for i, metric in enumerate(metrics):
plt.subplot(1, num_metrics, i + 1)
plt.plot(history.history[metric], label=f'Train {metric}')
val_key = 'val_' + metric
if val_key in history.history:
plt.plot(history.history[val_key], label=f'Val {metric}')
plt.title(f"{metric.capitalize()} Grafiği")
plt.xlabel("Epochs")
plt.ylabel(metric)
plt.legend()
plt.grid(True)
plt.tight_layout()
plt.show()
plot_history(history)
cm=confusion_matrix(y_true, predictions)
print("Confusion Matrix:\n", cm)
sns.heatmap(cm, annot=True, fmt='d', cmap='Blues', xticklabels=trainset.class_names, yticklabels=trainset.class_names)
plt.xlabel('Predicted Label')
plt.ylabel('True Label')
plt.title('Confusion Matrix')
plt.show()
print("\nClassification Report:\n", classification_report(y_true, predictions, target_names=trainset.class_names))
predictions = model.predict(testset)
predictions = predictions.flatten()
y_true = np.concatenate([y for x, y in testset], axis=0)
threshold = 0.5
predictions = (predictions > threshold).astype("int32")
plt.figure(figsize=(10, 10))
for images, labels in testset.take(1):
for i in range(9):
ax = plt.subplot(3, 3, i + 1)
plt.imshow(images[i].numpy().astype("uint8"))
plt.title(f"Pred: {predictions[i]}, True: {labels[i].numpy()}")
plt.axis("off")资料获取,更多粉丝福利,关注下方公众号获取


AtomGit 是由开放原子开源基金会联合 CSDN 等生态伙伴共同推出的新一代开源与人工智能协作平台。平台坚持“开放、中立、公益”的理念,把代码托管、模型共享、数据集托管、智能体开发体验和算力服务整合在一起,为开发者提供从开发、训练到部署的一站式体验。
更多推荐

 30
30 0
0- 0



 已为社区贡献4条内容
已为社区贡献4条内容

所有评论(0)