独家发现:近半数AI预测模型不如“算平均值”——这个开源平台终结了单细胞扰动预测的混乱局面
论文信息
标题:scArchon: a scalable benchmarking framework for assessing single-cell perturbation models
独家发现:近半数AI预测模型不如“算平均值”——这个开源平台终结了单细胞扰动预测的混乱局面
一句话速览: 海德堡大学团队开发了scArchon,一个全面、可复现的基准测试框架,系统评估了9种主流单细胞扰动预测模型。结果令人震惊:近半数模型表现不如简单的线性模型,且部分模型存在“生物幻觉”,生成看似合理但实际虚假的生物学结论。这项工作为个性化医疗中的药物响应预测设立了新的评估标准。
当AI谈“预测药效”时,它到底靠不靠谱?
想象一下:一位肿瘤医生拿到你的癌细胞样本,AI系统在几分钟内预测出哪种药物对你的特定癌细胞最有效——听起来像是科幻电影里的场景,但这正是单细胞扰动预测模型试图实现的目标。
问题是,目前市面上已经有十几种声称能完成这个任务的AI工具。它们有的用变分自编码器,有的用最优传输,还有的用生成对抗网络。每一篇论文都在展示自己模型如何完美地“预测”了药物处理后的细胞状态。但是,你真的能用这些模型信任它们输出的结果吗?
海德堡大学的研究团队意识到一个尴尬的事实:这个领域缺乏一个公正、全面、可复现的“裁判”。每种新模型发布时,往往只跟自己选定的几个对手在特定的数据集上比试,就像拳击手只在自己主场比赛,还挑对手。结果呢?这个研究发表在《Genome Biology》上的论文给出了令人清醒的答案。
隐藏的危机:为什么现有评估“骗”了我们
要理解这个问题的严重性,先来了解单细胞扰动预测任务在做什么。
科学家们有一个包含“对照组”和“处理组”(比如某种药物)的单细胞RNA测序数据。目标是训练一个模型,让它学会从“对照组”到“处理组”的映射关系。然后,给定一个全新的、从未见过的“对照组”细胞(来自不同患者或不同细胞类型),模型要预测出这个细胞在被药物处理后会变成什么样。这就是所谓的“分布外预测”。
听起来很直接,对吧?问题来了:每个团队都在用自己的规则玩游戏。
有的团队用主成分分析(PCA)降维来展示预测结果多么接近真实值;有的用UMAP;有的用t-SNE。不同的可视化方法竟然会给出相互矛盾的结论!在PCA中看起来完美重合的点云,切换到UMAP后却明显分离。这意味着,仅仅通过可视化“看起来不错”的评价方式,本质上是不可靠的。
更关键的是,大多数模型只报告一些宏观统计指标,比如均方误差(MSE)。但MSE低就代表模型学对了生物学吗?这个研究给出的答案是:未必。
scArchon:一个公平的裁判入场
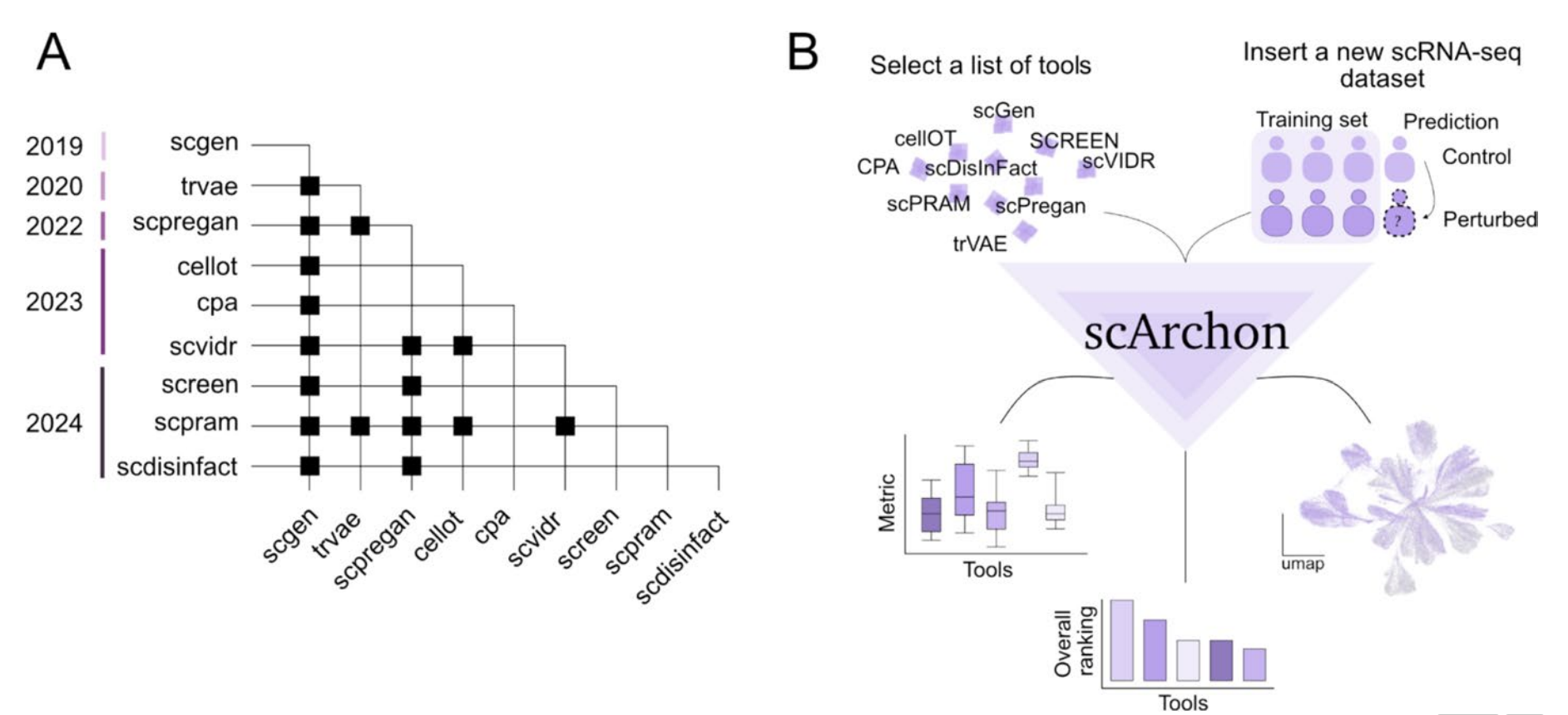
研究团队开发的scArchon是一个基于Snakemake构建的模块化基准测试平台。它的创新之处在于:
第一,容器化部署。 每个模型都被打包成Docker镜像,彻底解决了“我的环境能跑,你的跑不了”这种经典问题。这意味着任何人都可以复现实验结果。
第二,多维度评估。 不仅仅看宏观统计指标,还加入了生物学层面的评估——差异表达基因(DEG)的重叠度、基因集富集分析的相似度。这就好比考试不仅看总成绩,还要拆解每道题的得分。
第三,跨数据集验证。 研究团队在6个不同的数据集上进行了测试,包括人类PBMC、小鼠肠上皮细胞、胶质母细胞瘤患者样本等,覆盖了从简单到复杂的多种场景。
震撼的结果:近半数模型不如“算个平均差”
研究的结果可以用一个词来概括:始料未及。
团队定义了两个基线模型:
-
线性模型:直接计算所有训练细胞中,每个基因在处理前后的平均表达差异,然后将这个“平均变化”加到新细胞上。
-
控制基线:直接使用未处理的细胞作为“预测结果”(表示完全没有预测到任何效果)。
结果令人瞠目——在某些数据集上,CellOT、CPA、scPreGAN等工具的排名竟然低于线性模型,甚至接近控制基线。这意味着,这些复杂的深度学习模型花了几小时甚至几天训练,产出的结果还不如直接算个平均值有用。
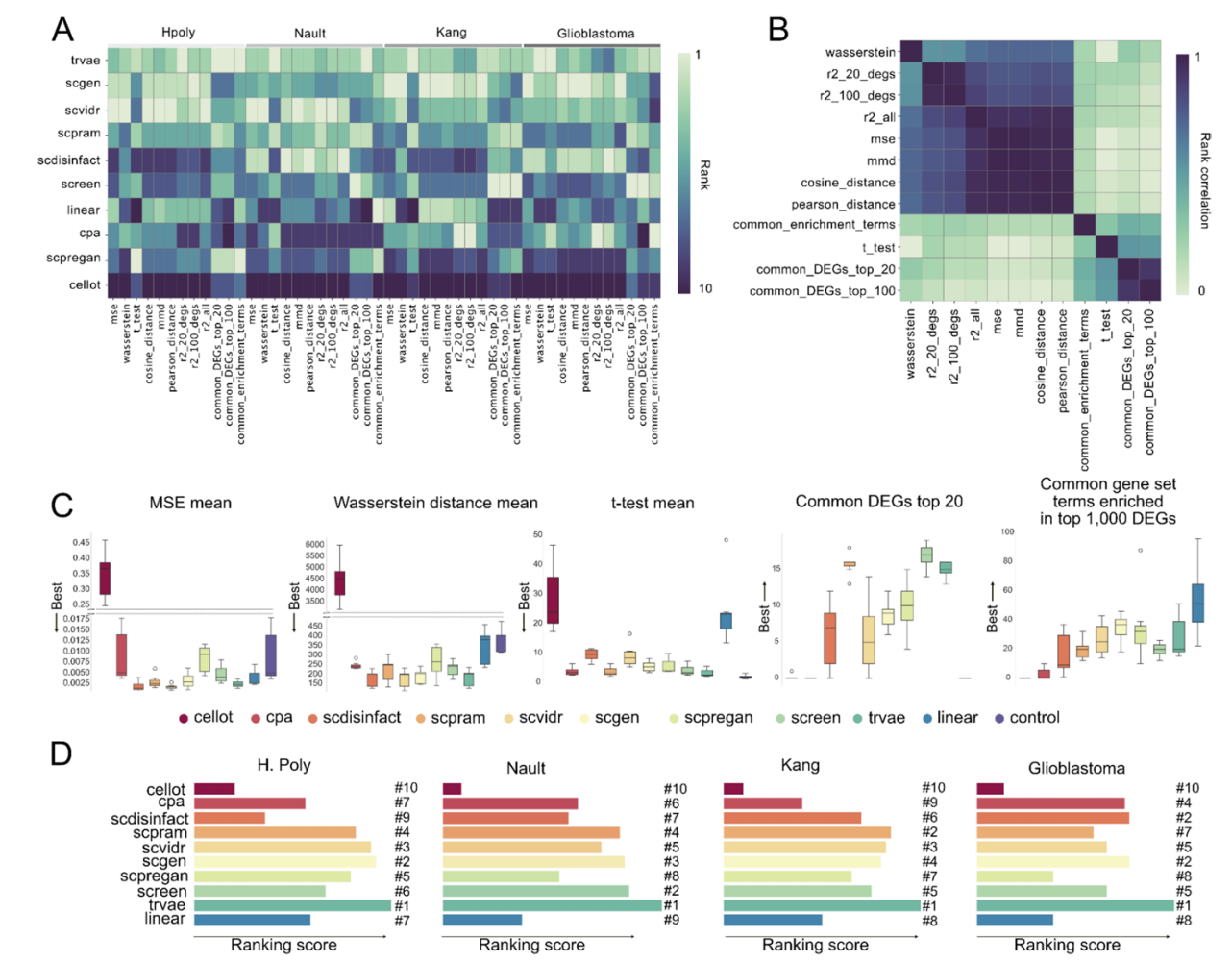
具体来看,trVAE、scGen、scPRAM和scVIDR在多个数据集上表现相对稳健。但即使是最好的工具,在面对更具挑战性的数据时(如胶质母细胞瘤患者样本,个体间差异极大),预测效果也大打折扣。
“AI幻觉”入侵生物学:生成虚假的生物通路
这是整个研究中最值得警惕的发现。
研究团队深入分析了模型预测结果的生物学意义。他们比较了预测细胞与真实扰动细胞中差异表达基因所富集的基因本体(GO)通路。
他们发现了令人不安的现象:某些模型在宏观指标上表现不错,但在基因层面却“胡编乱造”。
比如scGen模型预测CD4T细胞在干扰素处理后会激活252个GO通路,而真实扰动只激活了72个。其中那些预测特有的通路,经过检验发现其相关基因在预测细胞中的表达被人为抬高。研究团队将这种现象类比为大型语言模型中的“幻觉”——模型生成了看似合理但实际虚假的生物学结论。
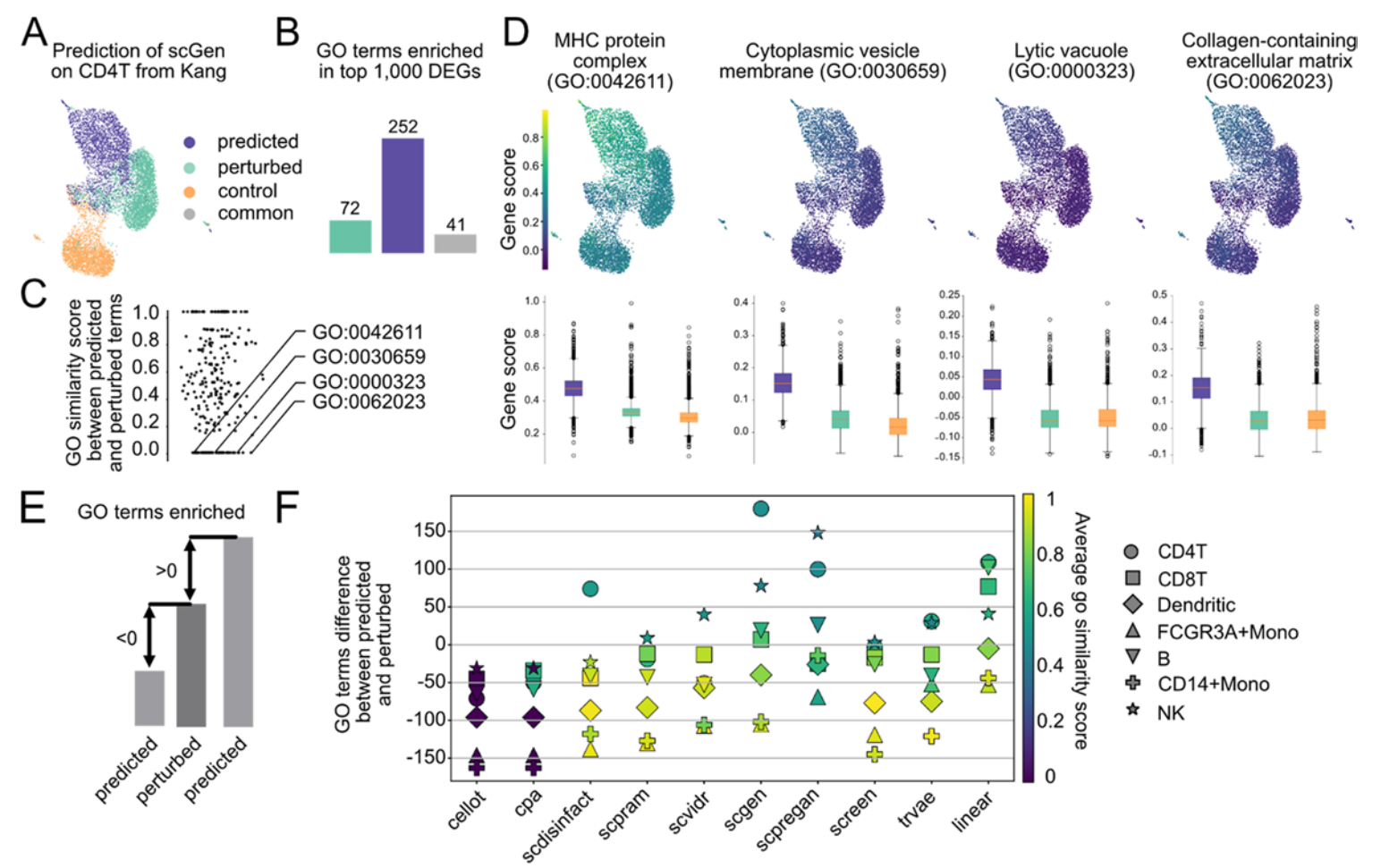
反过来,有些模型在宏观统计指标上得分不高,但在捕获差异表达基因方面却表现尚可。这说明,仅仅依赖MSE或R²这类指标来评判模型,可能会完全错失模型的真实表现。
跨物种翻译:当前的“不可能任务”
研究还测试了一个极具临床意义但极具挑战性的任务:跨物种预测。
具体来说,模型用小鼠细胞学习干扰素α的响应模式,然后预测人类细胞在同样条件下的响应。结果不出所料——所有模型都失败了。
即使是最先进的模型,在评估干扰素响应通路激活程度时,预测的信号通路活性分布与真实扰动的人类细胞存在显著差异(p<0.001)。这表明,当前的模型还远未能捕捉到跨物种保守的生物学机制,而这是个性化医学中将动物模型结果转化到人类应用的关键一步。
意义与展望:我们需要更务实的AI
这项研究的意义不仅在于“给模型打分”。它揭示了一个更深层的问题:我们可能对AI预测单细胞扰动的期望过于乐观。
预测每一个基因在每一种细胞类型中的完整表达变化,可能是一个“过于宏大的目标”。正如研究所指出的,或许更应该专注于预测特定关键信号通路的活性变化,或预测细胞的药物敏感性——这些更具体、更可验证的目标。
scArchon框架本身已成为开源工具,研究团队鼓励未来的方法开发者使用这个平台进行公正评估,并提供容器化的工具。这是推动该领域健康发展的关键一步——没有盲测的模型,不值得被信任。
局限性
当然,这项基准测试也有其局限。由于采用统一的通用评估框架,研究难以对所有数据集进行深入的生物学验证。某些特定基因的细微变化可能在宏观指标中被淹没。此外,研究以默认参数运行所有模型,而某些模型可能在精心调参后表现更好。最后,限于计算资源,最新的基于大语言模型的工具(如c2s)未被纳入主要基准比较。
看看当前的评估体系,再想想那些声称“AI即将实现精准用药预测”的宣传,我们是否应该停下来问一问:在单细胞生物学这个领域,当AI模型输出的漂亮数字背后可能隐藏着精心的挑选和虚假的信号时,我们该如何鉴别一个预测模型是否真正实用?是在更简单、更透明的基线模型面前证明自己的价值,还是在花哨的架构论文里迷失方向?

AtomGit 是由开放原子开源基金会联合 CSDN 等生态伙伴共同推出的新一代开源与人工智能协作平台。平台坚持“开放、中立、公益”的理念,把代码托管、模型共享、数据集托管、智能体开发体验和算力服务整合在一起,为开发者提供从开发、训练到部署的一站式体验。
更多推荐



 6
6 0
0- 0



 已为社区贡献16条内容
已为社区贡献16条内容

所有评论(0)