【文献阅读】化学空间边缘的分子深度学习
论文基本信息
-
标题:Molecular deep learning at the edge of chemical space
-
作者:Derek van Tilborg, Alisa Rossen, Francesca Grisoni
-
期刊:Nature Machine Intelligence, 2026年4月
-
核心贡献:提出基于重构损失的不熟悉度指标,联合训练分子自编码器和活性分类器,在高通量筛选库中发现了结构新颖(Tanimoto相似度仅0.28)且具有低微摩尔活性的分子。

一、背景:药物发现中的“分布外”难题
在药物发现中,我们常常面临一个窘境:深度学习模型在已知分子上表现优异,但一旦遇到结构全新的分子(即分布外,Out-of-Distribution,OOD),预测性能便会断崖式下跌。传统的分子相似性(如Tanimoto系数)或预测不确定度难以准确捕捉这种“陌生感”。
那么,有没有一种指标,能告诉模型:“这个分子我从来没见过,我的预测可能不准”?
2026年4月,van Tilborg, Rossen 和 Grisoni 在《Nature Machine Intelligence》上发表了一项题为 “Molecular deep learning at the edge of chemical space” 的工作,提出了一种全新的度量——不熟悉度(Unfamiliarity, 𝕌(x)),并结合联合分子模型(JMM),在大规模筛选库中成功发现了结构新颖且具有低微摩尔活性的分子。
二、核心思想:用“重建误差”量化陌生感
论文的核心思想,是构筑一个能够同时学习“分子结构”和“生物活性”的模型。它经历了先“打基础”再“专精”的两个主要阶段,其训练过程也包含了巧妙且严格的评估环节。
🧠 第一步:学习“分子语法”—— 预训练阶段
在这个阶段,模型的目标不是预测活性,而是学习分子的“内在结构规律”,即它们在化学上是否是“合理”的。
-
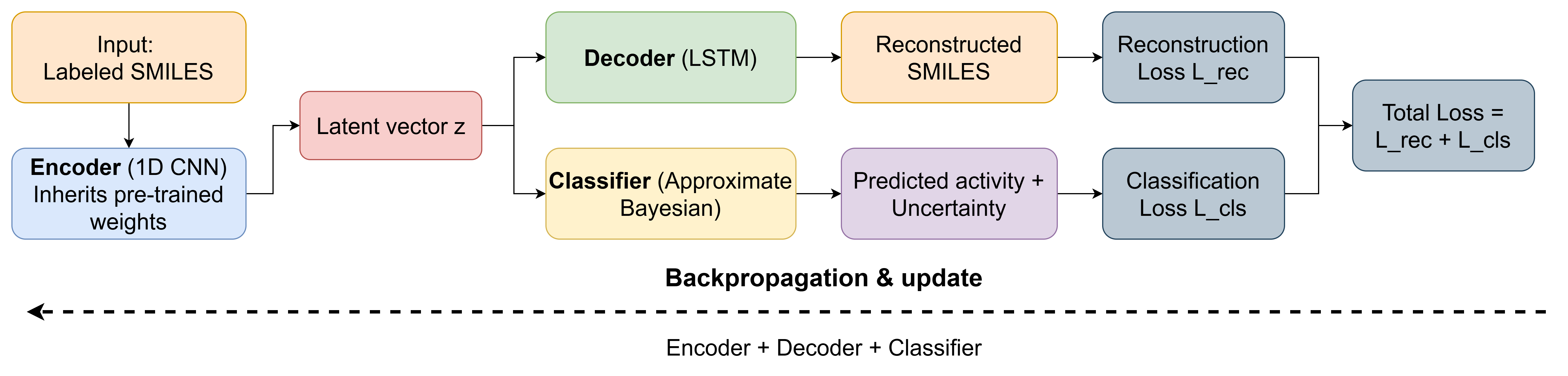
搭建骨架(Model Architecture):论文构建了一个称为联合分子模型 (JMM) 的半监督自编码器(Semi-supervised Autoencoder)。
-
编码器:它将分子的一维字符串表示(即SMILES符号序列, V=35)编码成一个压缩的潜在向量 (z),可以理解为模型对分子“结构”的抽象理解。
-
解码器:它负责将潜在向量
z解码回原始的SMILES字符串。这一步至关重要,因为它迫使模型必须忠实地捕获分子的结构信息。
-
-
海量学习:为了让模型充分学习“分子语法”,研究者使用了 ChEMBL数据库约120万个未标记的分子进行预训练。
🎯 第二步:学习“生物活性”—— 微调与联合训练阶段
当模型理解了分子的“语法”后,便可学习分子的“语义”,即其特定的生物活性。
-
新增分类器:在预训练好的
编码器之后,论文添加了一个近似贝叶斯分类器。它以z为输入,开始学习预测分子对特定靶点(如激酶PIM1)的生物活性,同时还能给出预测不确定性的评估。 -
联合训练(Joint Training):这是最终训练的关键。对于每一个有标签的分子,模型都会同时计算两个损失(Loss),并进行联合优化。
-
分类损失:评估其对生物活性的预测是否正确。
-
重构损失:衡量解码器还原SMILES的能力,可通过标准化负对数似然损失计算-31。该重构损失是模型衡量分子“熟悉度”的核心。
-
通过这种联合训练,模型的潜在空间 z 被迫同时编码结构信息和功能信息,形成了一个强大的分子表征。
🚀 第三步:定义“不熟悉度”并划分布局外数据(OOD)
训练完成后的JMM不仅能进行预测,还能对分子的“陌生度”打分,这有赖于特殊的训练/测试集划分。
-
定义“不熟悉度” (Unfamiliarity, 𝕌(x)):论文将“不熟悉度”定义为重构损失的对数
U(x) = log Reconstruction Loss(x)。意思是,模型对一个分子越“不熟悉”(如OOD分子),其解码器的重构效果就越差,重构损失越高,不熟悉度也就越高。 -
划分布局内(ID)/布局外(OOD)数据集:为了评估模型的泛化能力,论文并未采用传统的随机划分方法,而是采用前文提到的谱聚类来主动划分数据。
-
流程逻辑:先用所有数据计算出拉普拉斯特征值和谱嵌入。
-
肘部法则确定k:通过观察这些特征值,找到“肘部”,从而确定最佳聚类数(例如 k=11)。这代表数据天然形成了11个紧密的“族群”。
-
主动划分:接着,研究者将这11个“族群”中,那些距离其他所有族群最远的,即化学结构最独特的部分簇直接指定为OOD测试集(占~25%),其余的作为ID训练/测试集。通过这种方式保证了模型遇到的OOD分子是真正的结构“异类”。
-
-
分配新分子:对于一个全新的分子,其OOD归属可通过将它的嵌入与这11个族群的簇中心进行比较,来作最近邻判断。
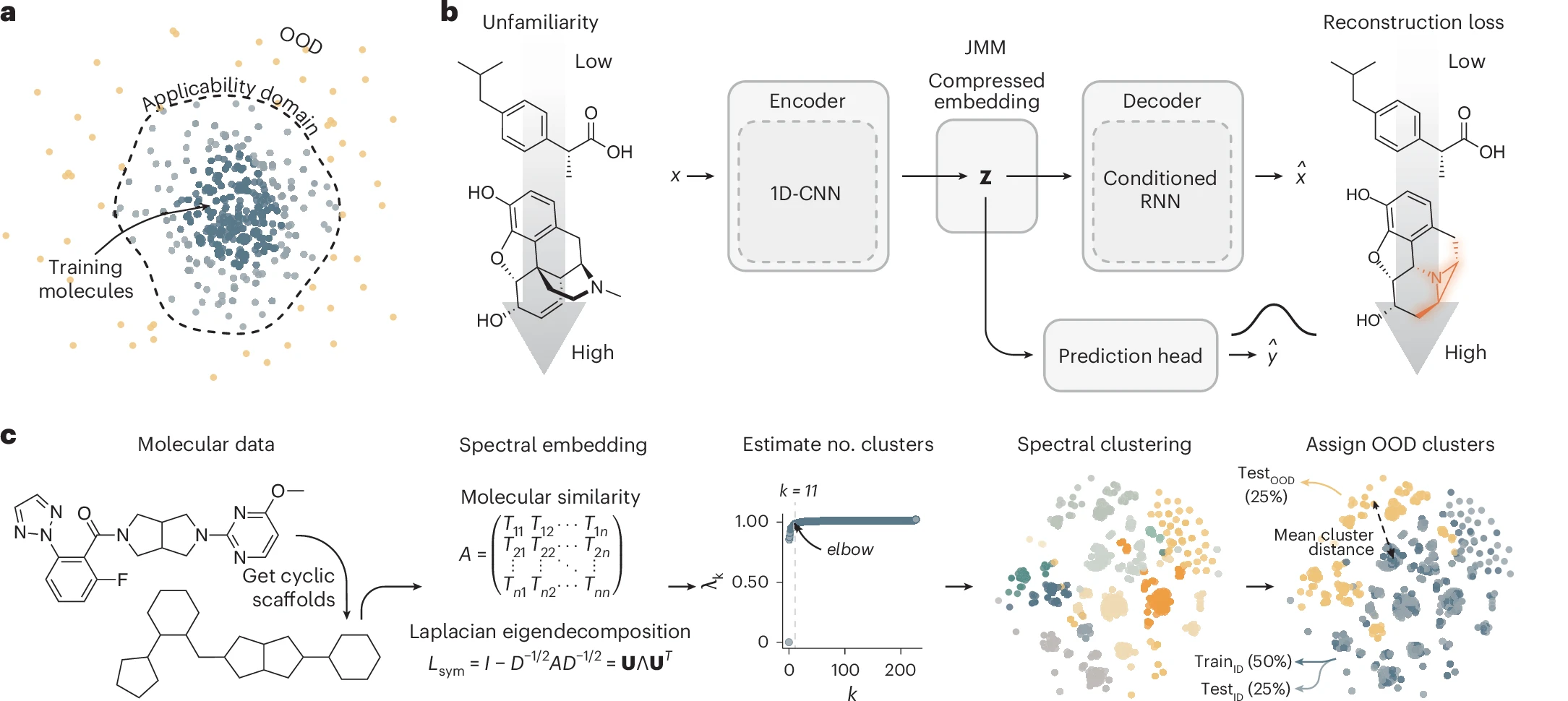
图1 使用联合模型估计分子数据的不熟悉度:
• a:适用域的概念性表示。在化学空间中,靠近训练数据的分子位于模型的适用域内,而位于该边界之外的分子则被视为分布外(OOD)样本。
• b:联合分子模型(JMM)的架构通过其重构损失来估计一个分子对模型的‘不熟悉’程度。CNN:卷积神经网络;RNN:循环神经网络。
• c: 通过谱聚类将分子数据划分为分布内和分布外两组,从而诱导分子分布偏移。
三、实验结果:不熟悉度凭什么“封神”?
3.1 不熟悉度的验证与优势
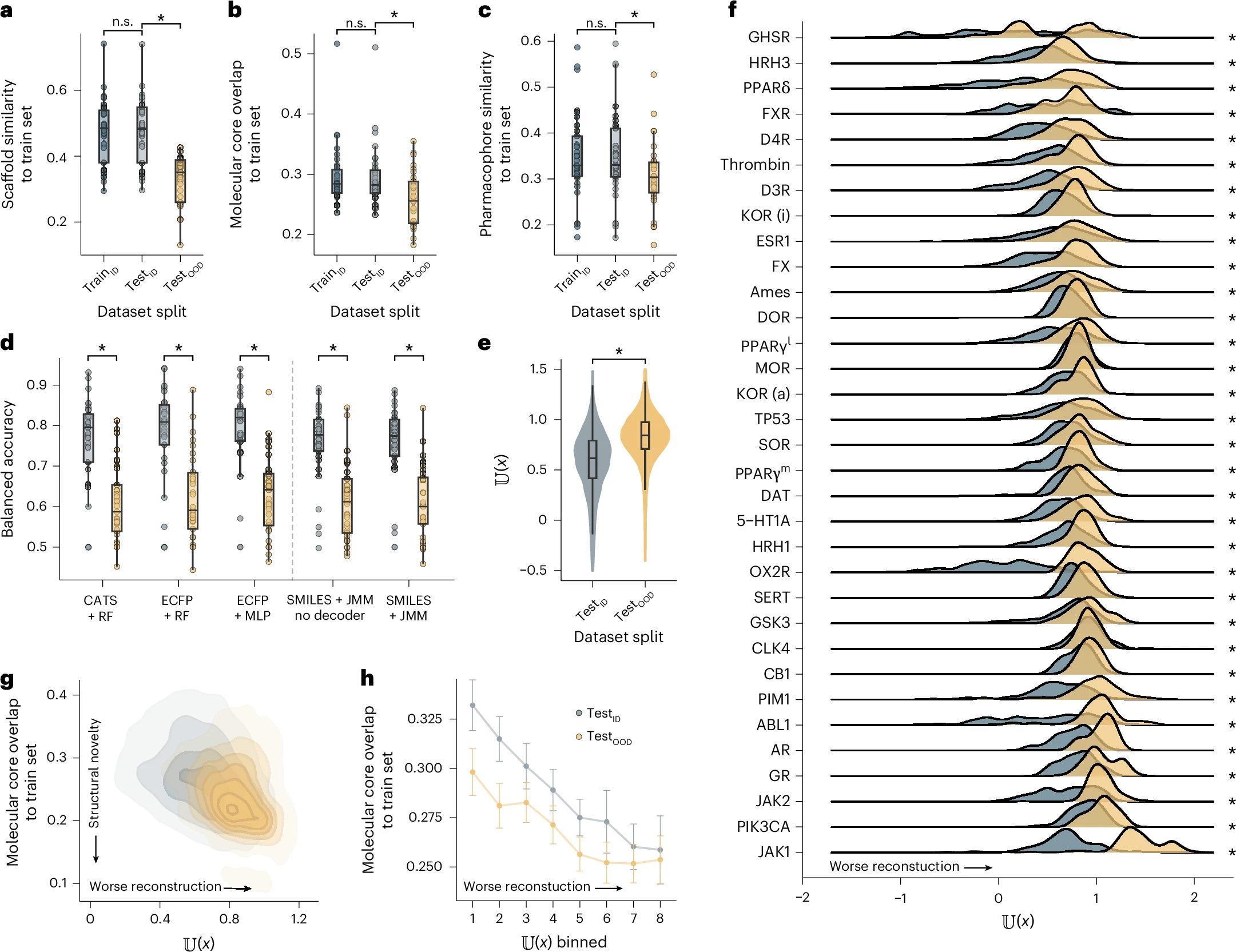
图2 系统地证明了不熟悉度的有效性和优越性。
-
a–c:验证数据集拆分的合理性。分别计算 trainID、testID、testOOD 与训练集的骨架相似性(a)、最大公共子结构分数 MCSF(b)、药效团相似性(c)。三项结果一致显示:testOOD 在三个化学维度上都显著偏离训练集(P < 0.05),说明谱聚类拆分是成功的。
-
d:比较不同模型的泛化能力。对比了 ECFP+RF、CATS+RF、普通MLP、无解码器的JMM、完整JMM 在 ID 和 OOD 上的平衡准确率。完整 JMM 在 OOD 上性能下降最小,且解码器的加入没有损害分类性能。
-
e–f:不熟悉度能否区分 ID 与 OOD?子图 e 将所有分子的 𝕌(x) 分布画在一起,testOOD 的 𝕌(x) 显著高于 testID(KS检验,P < 0.001)。子图 f 按33个数据集分别画箱线图,每个数据集中 testOOD 的 𝕌(x) 都明显更高,证明结论具有跨数据集的稳健性。
-
g–h:不熟悉度与结构陌生度的关系。横轴是 MCSF(与训练集的结构重叠度),纵轴是 𝕌(x)。散点图(g)和分箱柱状图(h)均显示强负相关:分子与训练集共享的核心片段越少(MCSF越低),不熟悉度越高。
3.2 虚拟筛选
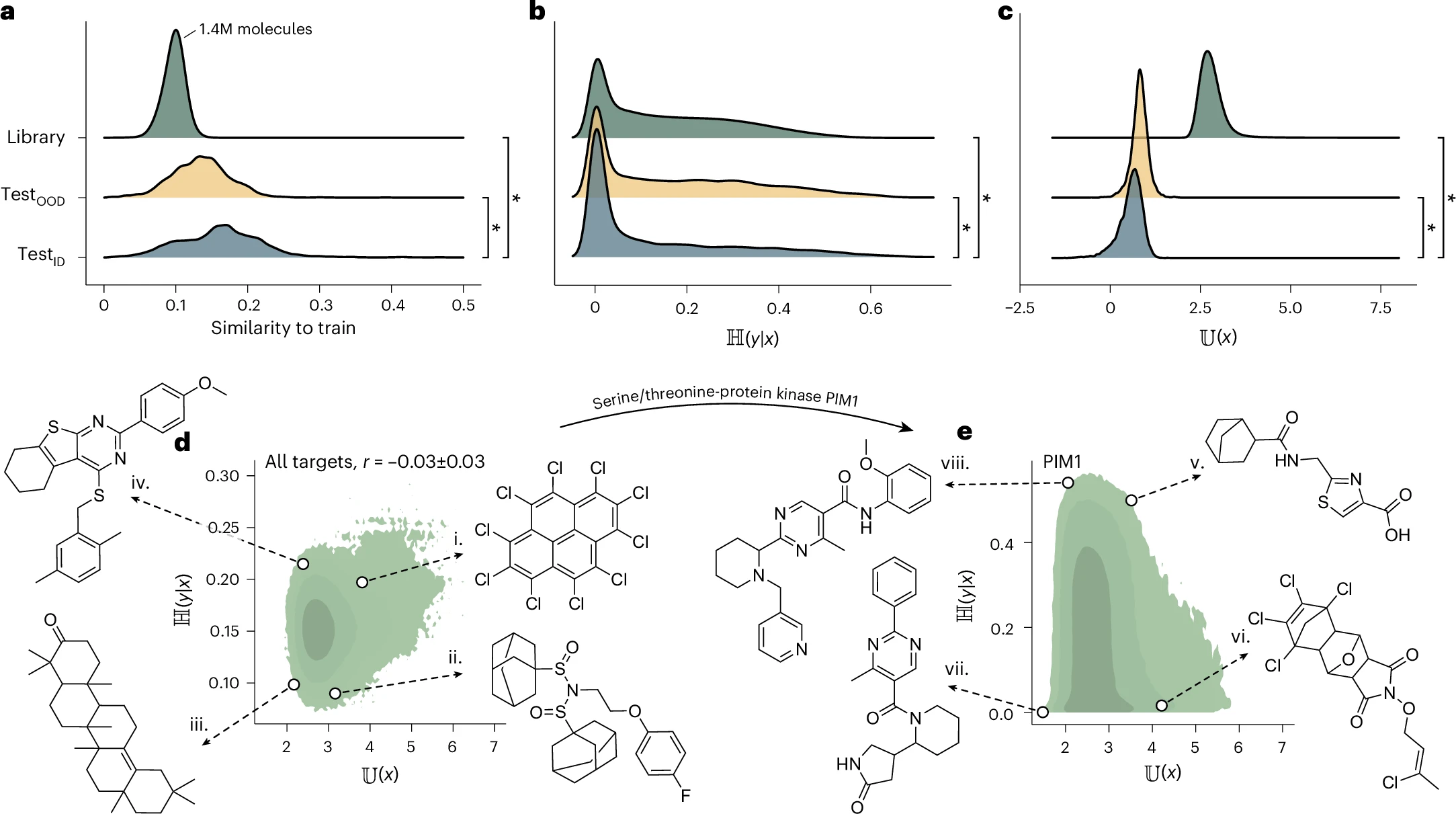
图3 针对140万个商业可得分子的虚拟筛选:
• a:来自testID(n=14,081)、testOOD(n=14,081)以及组合筛选库(n=46,048,926)中的分子,其ECFP指纹相对于各自训练集的平均Tanimoto相似度的分布。所有33个药物靶点的结果合并显示。
• b:testID、testOOD及组合筛选库中所有分子的预测不确定度 H(x) 的分布。
• c:testID、testOOD及组合筛选库中所有分子的不熟悉度分数 U(x) 的分布。
• d:筛选库中所有分子(n=1,395,422)的不确定度与不熟悉度之间的关系,数据为33个药物靶点的平均值。报告了平均斯皮尔曼相关系数 ± 标准误。标注了四个被预测对所有药物靶点均具有广泛生物活性的分子,每个分子都靠近理想点(例如,分子iii具有最低的不确定度和最低的不熟悉度;详见方法部分)。
• e:筛选库中所有分子针对丝氨酸/苏氨酸蛋白激酶PIM1(n=1,395,422)的不确定度与不熟悉度之间的关系。标注了四个被预测对PIM1具有生物活性的分子,每个分子均靠近理想点。
四、湿实验验证:真的能找到新颖活性分子吗?
理论验证再漂亮,不如湿实验一锤定音。作者从约18万个化合物的商业库中,根据预测不确定度与不熟悉度的三种组合,针对两个激酶靶点(PIM1 和 CDK1)各选出10个分子(总共60个),测试它们10 µM下的抑制活性。
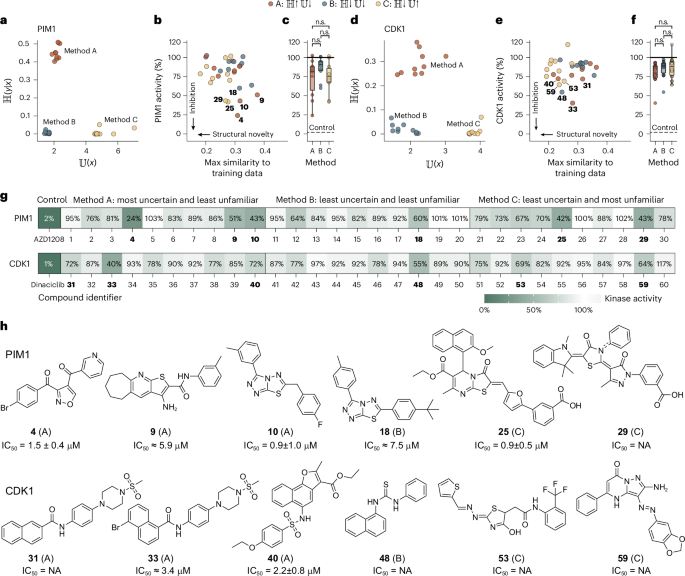
三种策略分别是:
-
A组:最高不确定度 + 最低不熟悉度(模型“犹豫但结构熟悉”)
-
B组:最低不确定度 + 最低不熟悉度(模型最舒适区,高置信度)
-
C组:最低不确定度 + 最高不熟悉度(模型自信预测的全新结构)
图3 解读:
-
a & d:展示 PIM1 和 CDK1 各组选出的分子编号。
-
b & e:每个分子的活性(越低越好)与它和训练集的最大 Tanimoto 相似度。C 组分子明显位于图中左侧(相似度低,大多 < 0.4),但仍有不少表现出低于 50% 的残留活性。
-
c & f:箱线图对比三组的活性分布。对 PIM1,C 组的中位数活性与 B 组接近,且显著优于 A 组;对 CDK1,C 组同样显著优于 A 组,与 B 组无显著差异。也就是说,C 组选出的高度陌生分子,其平均活性并不比舒适区的 B 组差。
-
g:将所有60个分子的活性值排序,粗体标识的分子进入下一步 IC₅₀ 测定。
-
h:最终展示6个最有前景的化合物的结构及其 IC₅₀ 值。其中多个 IC₅₀ 在低微摩尔范围,最低达到 0.64 µM(PIM1)。
湿实验室的结论非常有力量:不熟悉度最高的 C 组,尽管分子与训练集的平均 Tanimoto 相似度仅为 0.28±0.05,却依然能发现低微摩尔活性的新分子。这证明不熟悉度成功指引模型找到了真正的“化学空间边缘”活性分子,而单靠相似度或不确定度很难做到这一点。
五、代码模拟:手把手实现JMM的不熟悉度计算
为了帮助我自身更好理解的文章的核心思想,这里写了一个极简的PyTorch版本,使用随机生成的特征向量模拟分子。
import torch
import torch.nn as nn
import torch.optim as optim
import numpy as np
class JMM(nn.Module):
def __init__(self, input_dim, latent_dim, num_classes):
super().__init__()
self.encoder = nn.Sequential(nn.Linear(input_dim, 64), nn.ReLU(), nn.Linear(64, latent_dim))
self.decoder = nn.Sequential(nn.Linear(latent_dim, 64), nn.ReLU(), nn.Linear(64, input_dim))
self.classifier = nn.Sequential(nn.Linear(latent_dim, 32), nn.ReLU(), nn.Linear(32, num_classes))
def forward(self, x):
z = self.encoder(x)
x_recon = self.decoder(z)
logits = self.classifier(z)
return z, x_recon, logits
# 模拟数据:1000个分子,50维特征(例如ECFP降维)
input_dim, latent_dim, num_classes = 50, 20, 2
model = JMM(input_dim, latent_dim, num_classes)
optimizer = optim.Adam(model.parameters(), lr=1e-3)
recon_loss_fn = nn.MSELoss()
cls_loss_fn = nn.CrossEntropyLoss()
X_train = torch.randn(800, input_dim)
y_train = torch.randint(0, num_classes, (800,))
X_ood = torch.randn(200, input_dim) # 模拟OOD
for epoch in range(100):
z, recon, logits = model(X_train)
loss = recon_loss_fn(recon, X_train) + cls_loss_fn(logits, y_train)
optimizer.zero_grad()
loss.backward()
optimizer.step()
# 计算不熟悉度
model.eval()
with torch.no_grad():
_, recon_train, _ = model(X_train[:10])
_, recon_ood, _ = model(X_ood[:10])
unfamiliarity_train = np.log(recon_loss_fn(recon_train, X_train[:10]).item())
unfamiliarity_ood = np.log(recon_loss_fn(recon_ood, X_ood[:10]).item())
print(f"Train unfamiliarity: {unfamiliarity_train:.4f}, OOD unfamiliarity: {unfamiliarity_ood:.4f}")六、全文总结:三个层次的创新
| 层次 | 内容 | 意义 |
|---|---|---|
| 概念创新 | 提出“不熟悉度” = log(重构损失) | 简洁、可解释、与模型训练自然兼容 |
| 方法创新 | 联合分子模型(编码器+解码器+分类器) | 共享潜在空间同时学习结构与功能 |
| 实验创新 | 谱聚类主动拆分OOD + 湿实验验证 | 公正评估泛化性,并真正发现新颖活性分子 |
个人评价:
这项工作是“AI+药物发现”领域少有的把方法开发、统计学验证、湿实验闭环都做得非常扎实的研究。不熟悉度有望成为未来分子OOD检测的标准基线。
参考资料
van Tilborg, D., Rossen, A., & Grisoni, F. (2026). Molecular deep learning at the edge of chemical space. Nature Machine Intelligence.
https://www.nature.com/articles/s42256-026-01216-w

AtomGit 是由开放原子开源基金会联合 CSDN 等生态伙伴共同推出的新一代开源与人工智能协作平台。平台坚持“开放、中立、公益”的理念,把代码托管、模型共享、数据集托管、智能体开发体验和算力服务整合在一起,为开发者提供从开发、训练到部署的一站式体验。
更多推荐



 11
11 0
0- 0



 已为社区贡献4条内容
已为社区贡献4条内容

所有评论(0)