预测单突变如何重写蛋白质“社交网络“:eSIG-Net比你想象的更聪明
论文信息
标题:eSIG-Net: an interaction language model that decodes the protein code of single mutations
预测单突变如何重写蛋白质"社交网络":eSIG-Net比你想象的更聪明
一句话速览: 人类基因组中有数百万个意义不明的突变,但预测单个氨基酸改变如何影响蛋白质之间的相互作用(即"蛋白质社交网络")一直是个难题。eSIG-Net提出了一种全新的交互语言模型,仅从序列信息出发,就能以超过85%的准确率预测突变对蛋白质互作的影响,全面超越现有11种主流方法——包括需要耗费巨大算力的AlphaFold类结构方法。
背景与痛点:从"读密码"到"猜社交关系"的鸿沟
想象一下,你手里拿着一本由20种字母写成的天书——这就是蛋白质序列。每一个字母代表一种氨基酸。过去15年,基因组测序技术飞速发展,科学家们发现了海量的人类遗传变异。其中,错义突变(单个氨基酸被替换)是最常见的一类。
但问题来了:知道一个字母被换掉了,不等于知道这本书的整个故事情节会如何改变。
蛋白质很少单打独斗。它们通过与其他蛋白质结合来执行功能,形成一张庞大的"蛋白质社交网络"(蛋白质-蛋白质相互作用,PPI)。一个微小的突变,可能让原本亲密无间的两个蛋白质从此"绝交",也可能让互不相识的两个蛋白质突然"牵手"。
这就引出了计算生物学中的一个核心挑战——**"相互作用悬崖"问题**。就像化学中的"活性悬崖"现象(分子结构微小改变导致药效剧烈变化),单个突变往往引发蛋白质互作状态的大幅度、不可预测的改变。
在此之前,主流方法主要分两类:
-
基于序列的方法:用深度学习模型(如ESM、ProtT5)直接预测两个蛋白质是否结合。但它们通常把突变蛋白和野生型蛋白当作完全独立的样本处理,根本不去学习它们之间的差异。结果就是,面对"亲兄弟"(只有一个氨基酸不同)的两条序列,模型常常一脸茫然。
-
基于结构的方法:依赖AlphaFold等工具先预测蛋白质三维结构,再分析突变的影响。这种方法不仅计算成本极高(一次预测可能需要数小时甚至数天),而且面对单突变这种"微妙"的变化,AlphaFold预测的结构往往差异极小,根本无法区分。
简而言之,**此前的方法要么"看不见差异",要么"看得太贵"**。
核心方法:eSIG-Net如何破局?
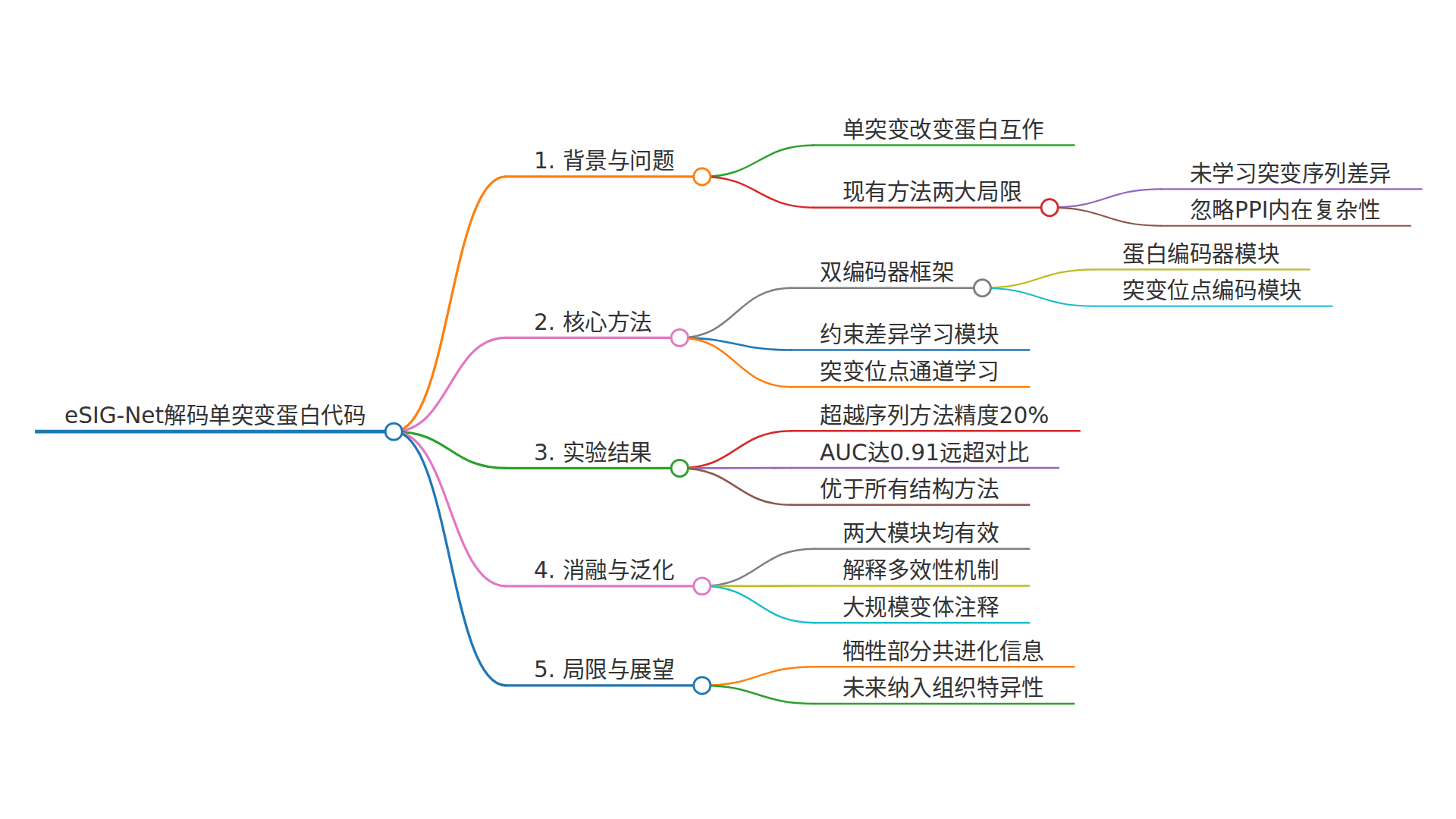
研究团队来自贝勒医学院、斯坦福大学、耶鲁大学等多个机构,他们提出的eSIG-Net(edgetic mutation sequence-based interaction grammar network) 设计了一个巧妙的"双通道"框架。
核心创新一:让模型学会"找不同"
传统的PPI预测模型输入的是"蛋白质A + 蛋白质B",输出"结合或不结合"。而eSIG-Net输入的是一个"三连体"(triplet):野生型蛋白 + 突变蛋白 + 互作蛋白。
模型要做的不再是"预测是否结合",而是预测突变前后的结合状态是否发生变化。这看似只是任务定义的微调,实则彻底改变了学习目标。
具体来说,eSIG-Net包含两个编码器模块:
-
PPI蛋白质编码器:分别获取"野生型-互作蛋白"和"突变蛋白-互作蛋白"的合并编码,然后送入一个约束差异模块。这个模块被设计成"放大镜",专门用来捕捉两个合并编码之间微妙的差异。
-
突变位点编码器:这里使用了蛋白质语言模型(ESM-2)提取残基级别的嵌入向量。但与传统方法不同的是,eSIG-Net只提取突变位点本身的嵌入,而不是整条序列。这样做的目的是让模型聚焦于最关键的变化,而不是被全局信息"稀释"。
核心创新二:约束差异学习
这是整个框架的"灵魂"。研究团队设计了一个特殊的损失函数:
ℒcd = (1/n(n-1)) Σ Σ (di/(1+λ×ci) - dj/(1+λ×cj))²
大白话解释:模型希望让"发生了互作改变"的样本对(c=1)之间的嵌入距离(d)被"拉开",而"没发生改变"的样本对(c=0)之间的嵌入距离被"压缩"。同时,通过联合训练原始PPI预测任务,避免了"把所有距离都变成0"的平凡解。
这种设计类似于让模型学会一种"语法"——单突变如何重写蛋白质互作的"句子结构"。
此外,团队还引入了对比学习机制,进一步拉大同类别与异类别样本的特征距离,让模型在面对高度相似的序列时也能做出精准判断。
实验结果:全面碾压,数据说话
在疾病突变数据集上的表现
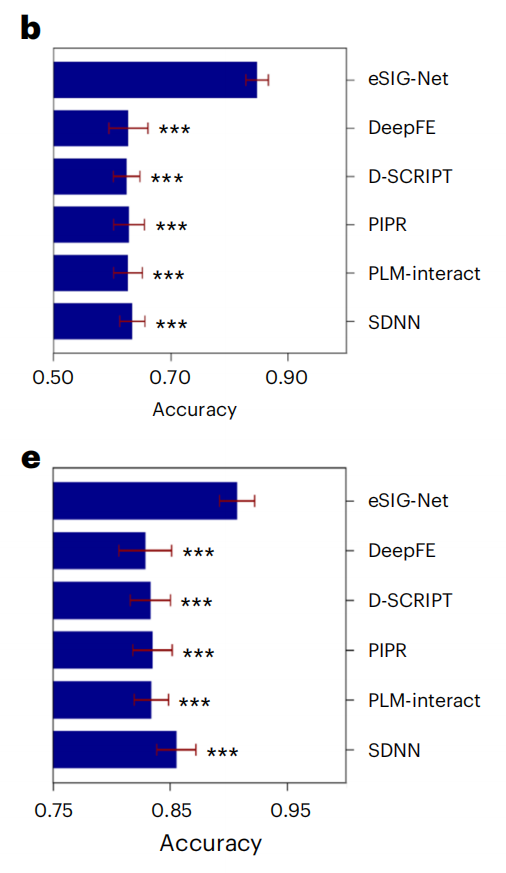
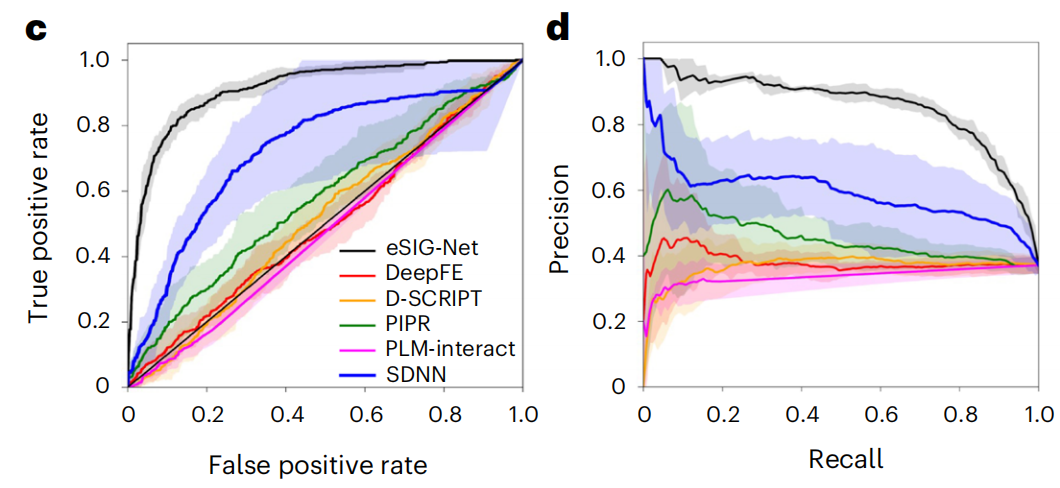
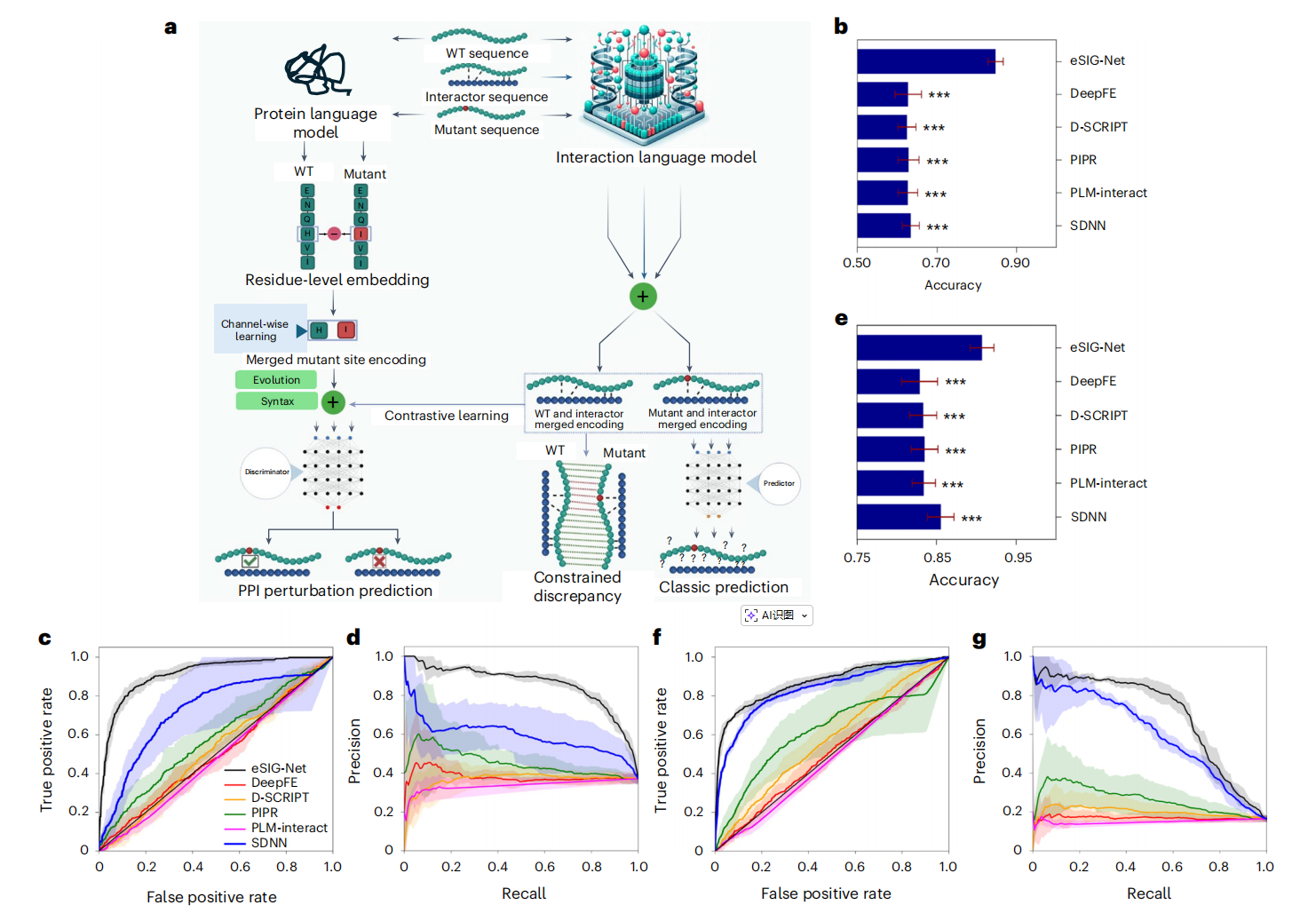
在包含1,633个PPI样本的疾病突变数据集上,eSIG-Net的表现令人瞩目:
-
准确率:eSIG-Net达到 0.85 ± 0.02,而最佳对比方法SDNN仅为0.63 ± 0.02,提升超过20%
-
AUC值:eSIG-Net高达 0.91 ± 0.02,第二名SDNN仅0.73 ± 0.15
-
平均精确率:eSIG-Net为 0.86 ± 0.01,对比方法最高也只有0.61
所有统计检验的p值均小于0.001,差异极其显著。
关键看点是:即使是目前公认最好的PPI预测模型PLM-interact和D-SCRIPT,在这项任务上的表现也几乎是"随机水平"(AUC在0.48-0.51之间)。这说明传统方法确实不具备处理单突变"细微差异"的能力。
在人群变异数据集上的表现
面对来自gnomAD数据库的近4,020个样本(其中只有约16%是阳性样本,极不平衡),eSIG-Net依然展现出强大的泛化能力:
-
准确率:0.90 ± 0.01
-
AUC:0.93 ± 0.01
相比之下,所有对比方法的准确率均未超过0.78。
消融实验:每个模块都很重要
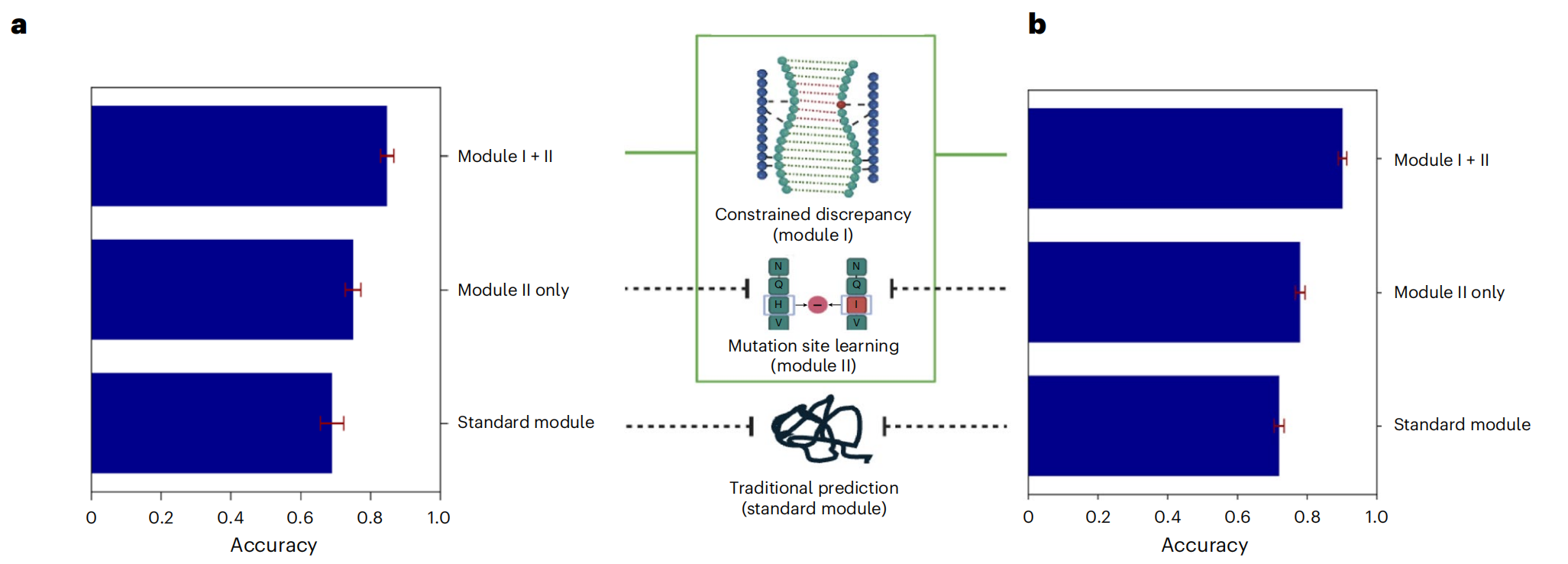
研究团队逐步拆解了eSIG-Net的各个模块:
-
标准模型(直接用ESM池化):准确率0.69
-
加入突变位点编码模块后:提升至0.75
-
再引入约束差异学习模块后:跃升至0.85
每一步都是实打实的提升,证明了设计的必要性。
对比结构方法:AlphaFold也甘拜下风
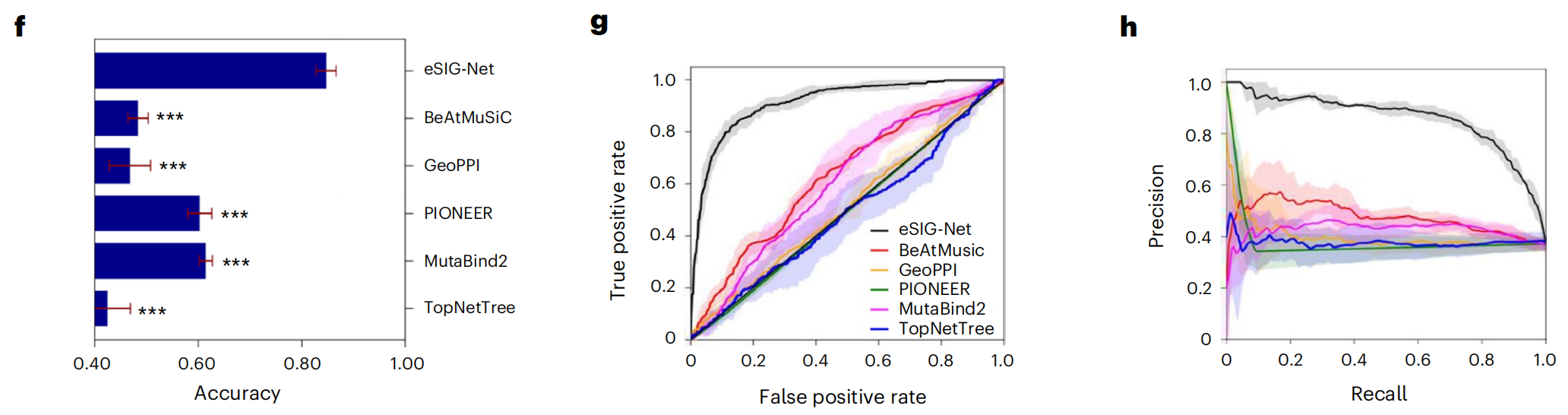
更令人意外的是,即使与需要输入蛋白质复杂三维结构的AlphaFold派方法(MutaBind2、BeAtMuSiC、GeoPPI等)相比,eSIG-Net依然全面胜出。这些结构方法的准确率普遍在60%或以下,而eSIG-Net的AUC高达0.91。换句话说,仅靠蛋白质的序列信息,eSIG-Net比那些需要先"猜结构再算变化"的方法准确得多。
用研究团队的话说:"目前最先进的结构方法FoldDock甚至无法预测某些疾病突变对互作的影响。"
真正的"读心"能力:解释多效性现象
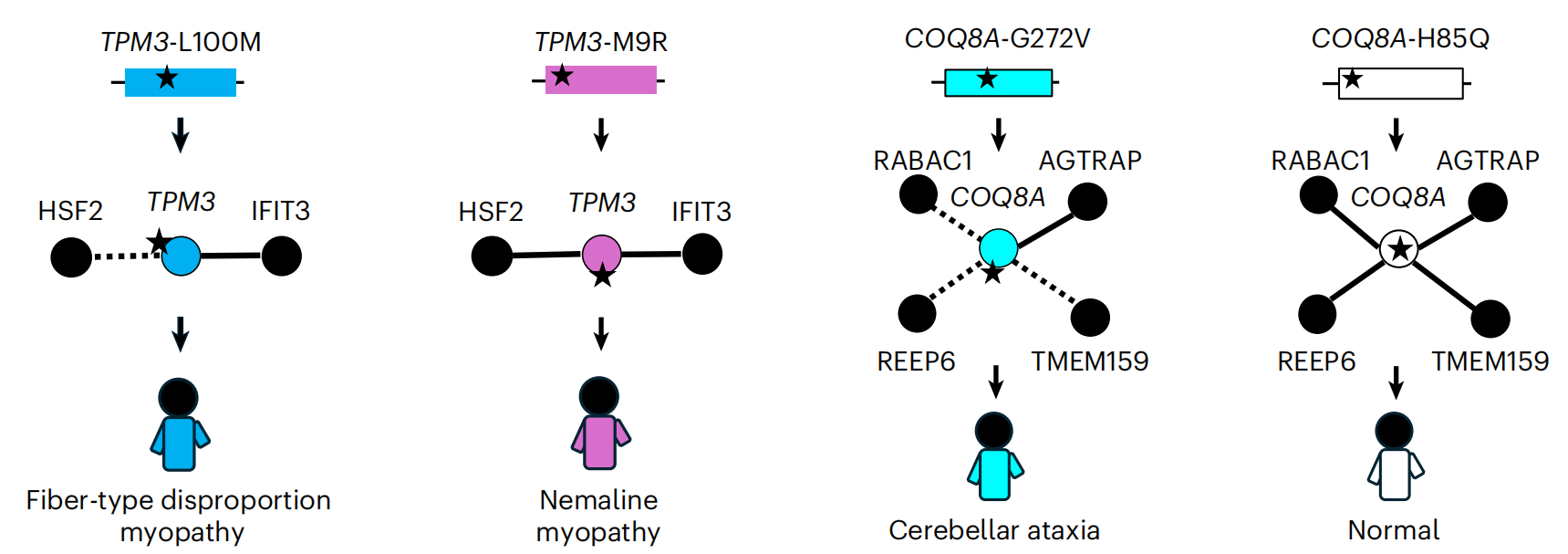
最能体现eSIG-Net价值的,是它对多效性现象的解释能力——同一个基因的不同突变,为什么会导致完全不同的疾病?
以TPM3基因为例:
-
L100M突变导致"纤维型不成比例肌病"
-
M9R突变导致"线虫样肌病"
eSIG-Net预测:L100M会选择性破坏TPM3与HSF2蛋白的互作(即"边缘性"扰动),而M9R则保留这种互作。HSF2恰好是肌肉发育和再生中的关键蛋白。这个预测完美解释了为什么两个突变虽然在同一基因上,却导致截然不同的临床表现。
另一个例子是COQ8A基因:H85Q(与其他疾病相关)和G272V(人群变异)也被eSIG-Net准确区分出完全不同的互作扰动模式。
意义与展望:VUS不再"未知"
人类基因组中,有数百万个被分类为"意义不明的变异"(VUS)。传统的实验验证方法(如深度突变扫描、功能变组学)虽然精确,但耗时费力。eSIG-Net提供了一种大规模计算筛选的替代方案。
这意味着:未来或许只需几分钟,就能对一个新发现的基因突变进行"体检",预测它可能扰乱了哪些蛋白质互作,从而快速锁定潜在的致病机制。
对于精准医学、药物靶点发现,尤其是癌症变异的功能注释,这项技术有巨大的应用前景。
局限性:清醒的自我审视
研究团队也坦诚指出了eSIG-Net的局限性:
-
牺牲了协同进化信息:目前版本使用基于序列的嵌入,没有引入多序列比对(MSA)信息。这虽然加速了计算,但在某些特定生物学背景下可能丢失宝贵的进化约束信息。
-
未考虑组织特异性:许多致病突变只在特定组织中发挥作用。eSIG-Net目前预测的是"普遍的"物理互作变化,但一个突变在心细胞和肝细胞中的影响可能完全不同。
-
从互作变化到疾病因果,仍有距离:模型告诉你"这个突变破坏了这个互作",但并不意味着这个互作的破坏就是疾病的原因。因果关系的确立需要更多的生物学验证。
写在最后
eSIG-Net的出现,标志着计算生物学从"预测结构"到"理解变化"的一个关键转折。它证明了:即使不依赖昂贵的结构数据,仅用序列信息和精心设计的差异学习策略,也能解码单突变对蛋白质社交网络的重写规则。
但这也引出一个更深层的问题:当AI已经能如此精准地预测分子层面的变化后,我们是否准备好理解这些变化在人体内——那个由数万个蛋白质、无数种细胞类型、复杂的组织微环境构成的真实系统里——究竟意味着什么?也许比预测突变更难的事,是理解突变在整个人体交响乐中的回响。你觉得呢?

AtomGit 是由开放原子开源基金会联合 CSDN 等生态伙伴共同推出的新一代开源与人工智能协作平台。平台坚持“开放、中立、公益”的理念,把代码托管、模型共享、数据集托管、智能体开发体验和算力服务整合在一起,为开发者提供从开发、训练到部署的一站式体验。
更多推荐



 8
8 0
0- 0



 已为社区贡献16条内容
已为社区贡献16条内容

所有评论(0)