700倍参数碾压巨头:这个RNA基础模型用进化“对比学习”重新定义了“少即是多”
论文信息
标题:Orthrus: toward evolutionary and functional RNA foundation models
700倍参数碾压巨头:这个RNA基础模型用进化“对比学习”重新定义了“少即是多”
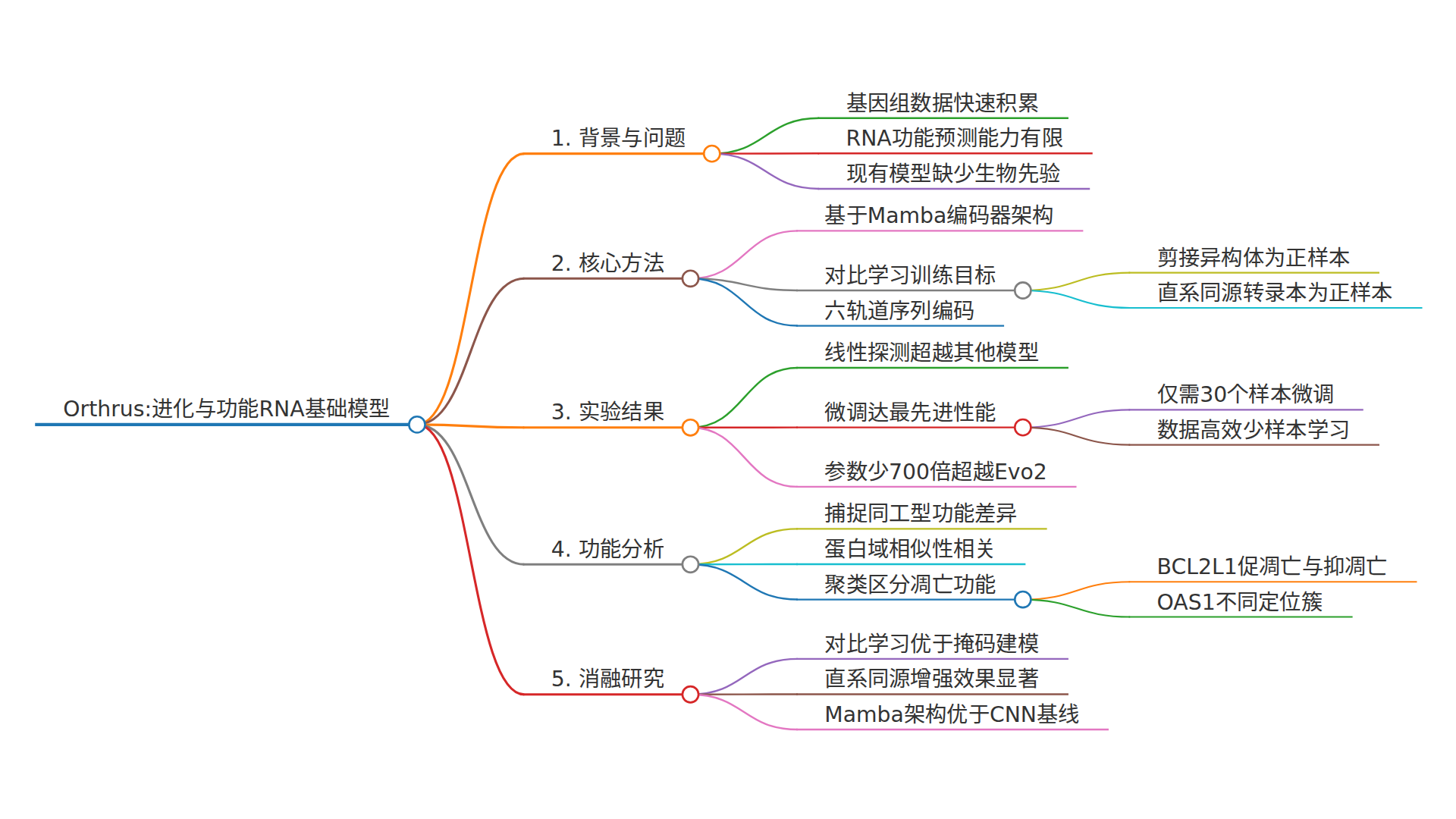
一句话速览: 现有基因组基础模型,像GPT或BERT一样,通过预测被遮罩的核苷酸或“下一个碱基”来学习DNA语言,却忽略了生物学中最重要的信号——进化。这篇论文提出的Orthrus模型,通过对比学习让模型学会识别功能相关的RNA序列(如剪接异构体和跨物种同源转录本),只用Evo2模型1/700的参数,就在mRNA特性预测任务上全面超越了现有方法,甚至在数据量低至30条样本时也能表现出色。
当大模型“看不懂”生物:一场关于“有效信息”的错配
过去几年,一场“暴力美学”的狂欢席卷了AI for Science领域。正如当年用GPU训练数百亿参数的语言模型一样,科学家们也将Transformer架构原封不动地搬到了基因组上,试图用海量的数据“喂”出一个能理解生命密码的“究极模型”。Evo、Nucleotide Transformer、DNABERT……一个比一个庞大,一个比一个昂贵。
然而,这些模型在预测最基础的mRNA性质时,表现却常常令人失望——它们能读懂序列,却“看不懂”哪段序列重要。
为什么?论文的作者们一针见血地指出了问题的核心:自然语言和基因语言的根本不同。
在自然语言中,“我爱你”如果改成“我恨你”,意义完全反转,每个词都承载着明确的信息。但在基因组中,人类30亿个碱基对里,只有大约2%是编码蛋白质的。剩下高达90%的基因序列,即便发生突变,对个体可能也毫无影响。如果我们用训练GPT的方法去训练基因组模型,模型会花大量精力去学习那些“无意义”的非编码区“噪音”,而真正关键的、决定mRNA稳定性和功能的“信号”,却淹没在庞大的背景噪声中。
这就像让一个学生去读一本几百万页的教材,但其中只有几百页是考点。如果他用“预测下一页内容”的方式来学习,他可能会成为一个出色的“翻页机器人”,却永远不知道考什么。
这个问题,在剪接异构体(splice isoforms)面前变得尤为突出。一个基因可以剪接出多种mRNA版本,它们共享绝大部分序列,却可能执行完全不同、甚至截然相反的功能(例如促进或抑制细胞凋亡)。传统模型很难区分这些“孪生”兄弟。
正是在这个背景下,来自多伦多大学、Vector Institute和纪念斯隆凯特琳癌症中心的研究团队,在《Nature Methods》上发表了一项堪称“四两拨千斤”的工作——Orthrus。
进化是最好的老师:用功能相似性替代“猜词游戏”
Orthrus的核心理念极其优雅:别让它记单词,让它认亲戚。
它抛弃了主流模型使用的“掩码语言建模”(你猜我遮了什么)或“下一词预测”(你猜下一个是什么),转而使用对比学习(Contrastive Learning)。
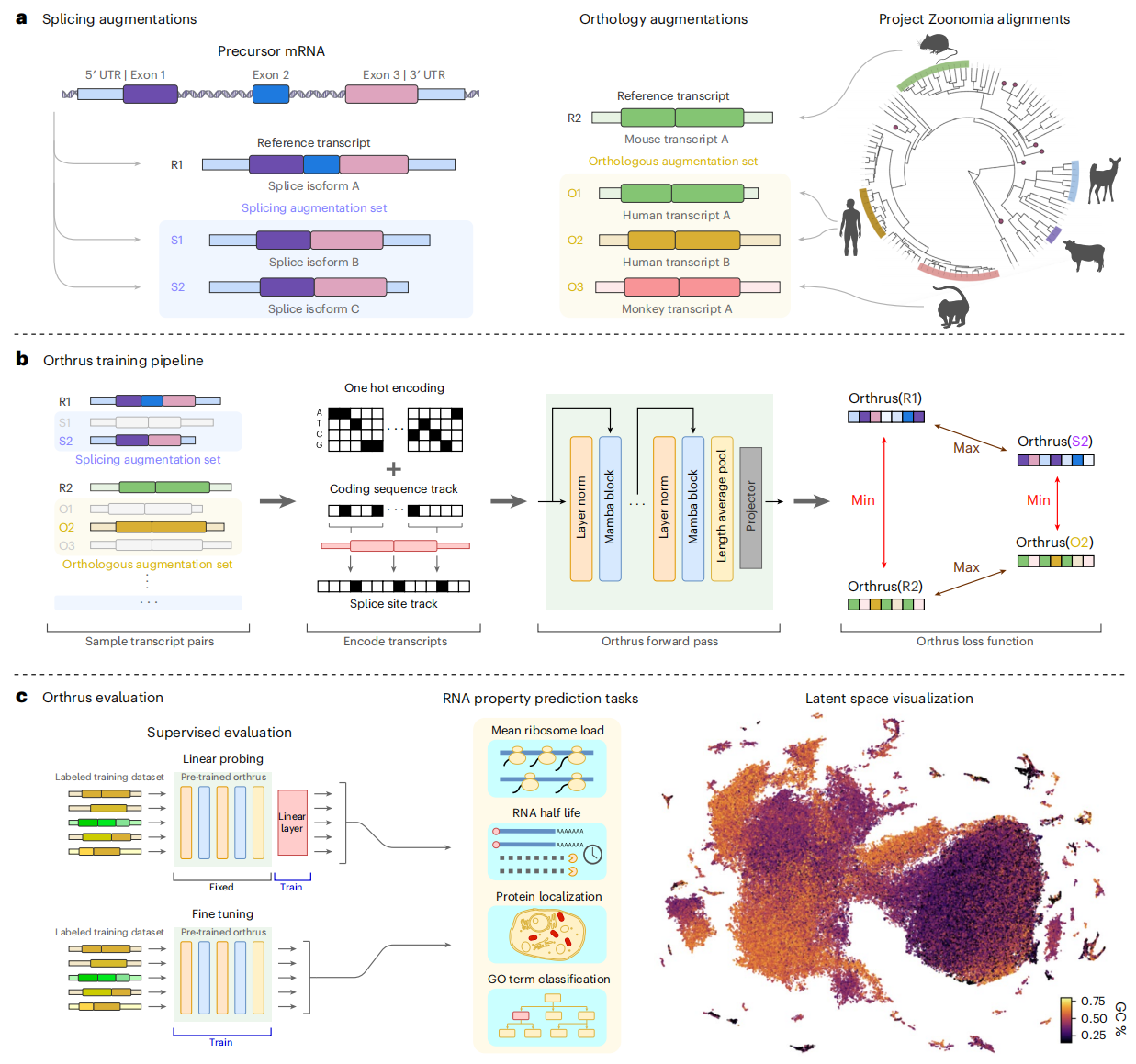
对比学习的逻辑很简单:给模型看两张图片,告诉它“这两张是猫”和“这两张不是猫”,让它学会提取“猫”的本质特征,忽略背景、光线等干扰。Orthrus正是借鉴了这一思想,但它用的不是图片,而是mRNA序列。
它的训练数据正对(positive pairs)来自两个极具生物学洞察的源头:
-
剪接异构体(Splicing Isoforms):同一个基因通过可变剪接产生的不同mRNA版本。虽然它们序列有差异,但通常仍共享核心功能。这迫使模型学习:哪些序列片段是决定功能的“不变项”。
-
直系同源转录本(Orthologous Transcripts):来自不同物种(如人类和小鼠、甚至400多种哺乳动物)的同源基因转录本。在动物界,一个基因如果想保存重要功能,必然不会乱变。这些跨物种的序列变体,就是进化本身设下的天然“数据增强”。
通过最大化这些正对的嵌入向量(embedding)相似度,同时推远其他所有不相关的序列,Orthrus学会了在潜在空间中将功能相似的RNA聚在一起。换句话说,模型不再需要费力地理解孤立的核苷酸,而是学会了“物以类聚”的生物学规则。
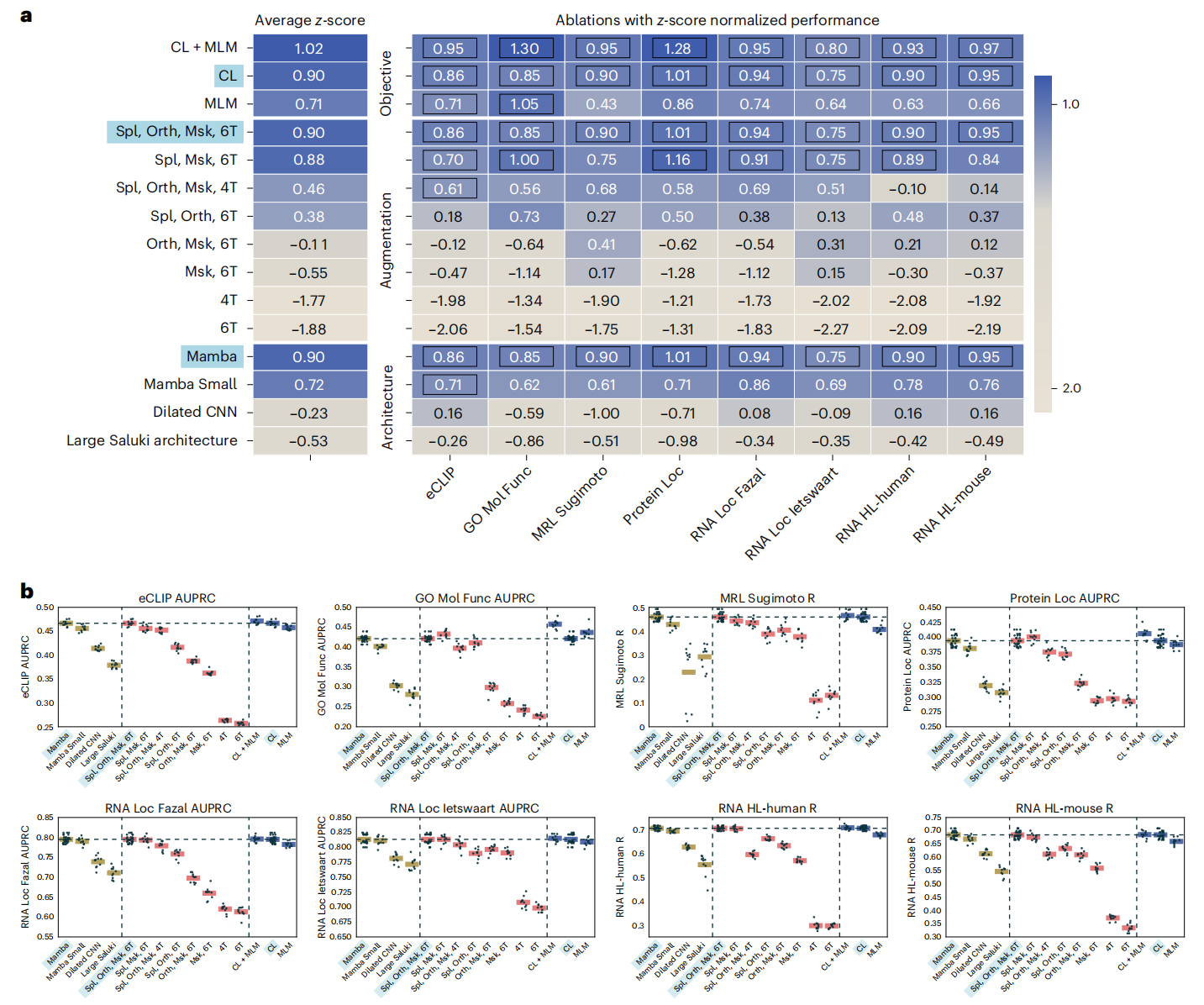
论文的消融实验(图3)清晰地展示了这一点:单独使用对比学习的效果(Z-score 0.90)远超仅使用掩码语言建模(0.71)。而将两者结合(CL+MLM),还能进一步提升(1.02)。更关键的是,引入同源序列(Orthology)带来的性能提升,远大于简单的序列掩盖(masking)。
只用Evo2的0.14%参数,它如何成为“全能选手”?
如果大模型的逻辑是“大力出奇迹”,Orthrus的逻辑则是“好钢用在刀刃上”。它选择了Mamba架构,一种能线性扩展内存的“选择性状态空间模型”,这比Transformer在处理上万碱基的长RNA序列时高效得多。但真正让它脱颖而出的,是那个带极强生物学先验的预训练目标。
结果令人惊叹。在mRNA性质预测的线性探针(linear probing)基准测试中,Orthrus展现出了统治级的性能:
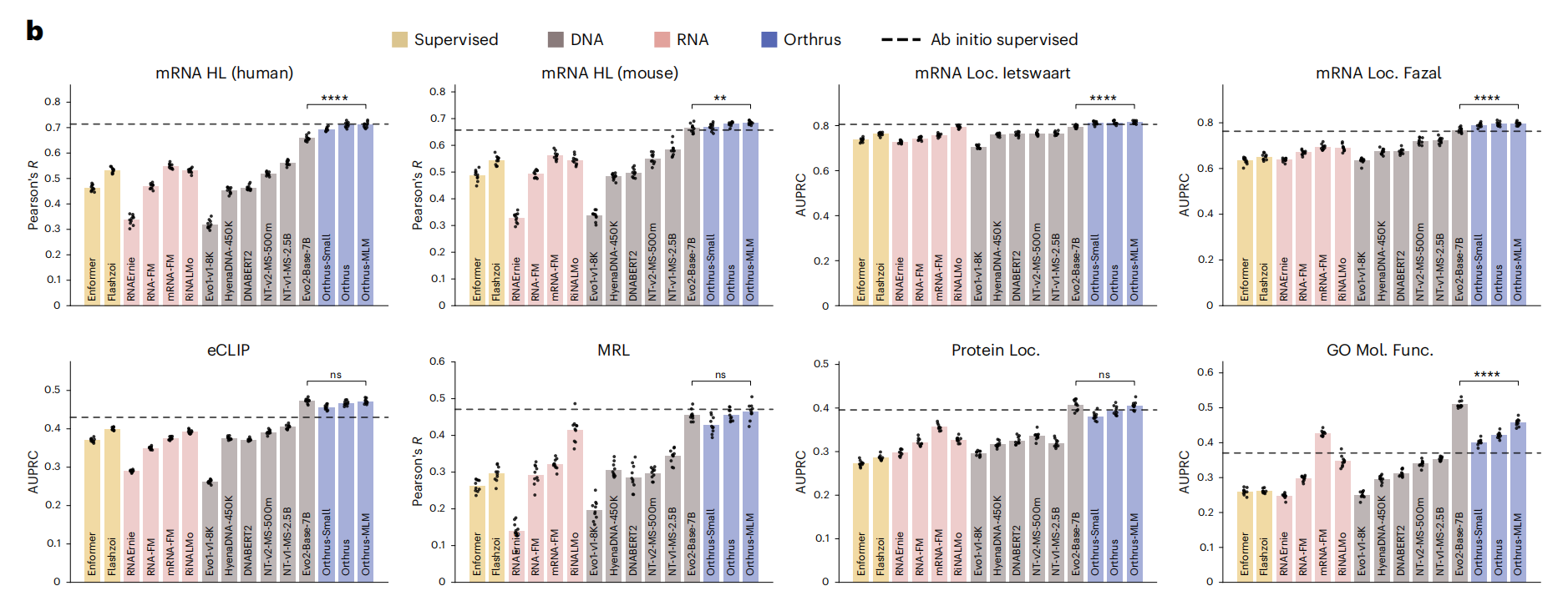
-
以少胜多:最强的Orthrus变体(10M参数)在7个任务上表现优于或打平了拥有70亿参数的Evo2。这是700倍的参数效率提升。Evo2包含一个70亿参数的模型,而Orthrus只有1000万参数。
-
超越“定制化”专家:更令人震惊的是,仅使用线性回归模型在Orthrus的固定嵌入上训练,就能击败或持平专门为这些任务定制的监督学习模型(如Saluki)。这意味着,Orthrus的嵌入本身就已经蕴含了丰富的功能信息,不需要后续再做复杂的网络设计。
-
“小数据”王者:在仅有30条有标签样本的极端少样本场景下,微调后的Orthrus在人类mRNA半衰期预测任务上,其皮尔逊相关系数能达到0.74,而从头训练的监督模型仅有0.53。这为实验数据稀缺的领域(如罕见病)提供了巨大可能性。
那么,Orthrus到底“看”到了什么?
为了验证模型的表征是否真的“理解”了功能,研究者们进行了一个精彩的分析(图4、图5)。
他们比较了同一基因不同剪接异构体的编码序列(CDS)重叠度和Orthrus嵌入相似度。结果发现,两者呈正相关,但有趣的是,出现了大量的“异类”:
-
IQCF6基因:两个异构体CDS重叠度高达0.84,但Orthrus相似度极低(0.29,处于1.52%的倒数位置)。AlphaFold3蛋白质结构预测显示,它们的蛋白质结构RMSD高达16.1Å(角秒),几乎完全不同。模型准确捕捉到了序列相似但功能迥异的“悖论”。
-
PANK2基因:CDS重叠度只有0.58,但Orthrus相似度非常高(2.30,处于55%的领先位置)。其蛋白质预测结构RMSD仅有0.55Å,几乎一模一样。模型成功识别了序列不同但功能高度相似的“隐形双胞胎”。
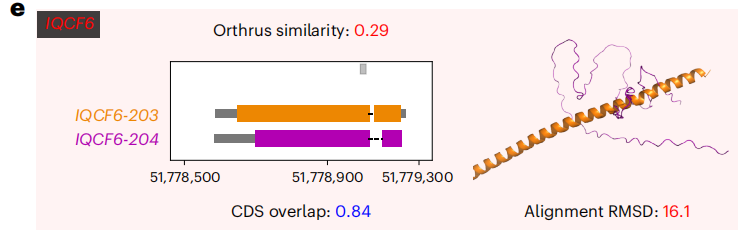
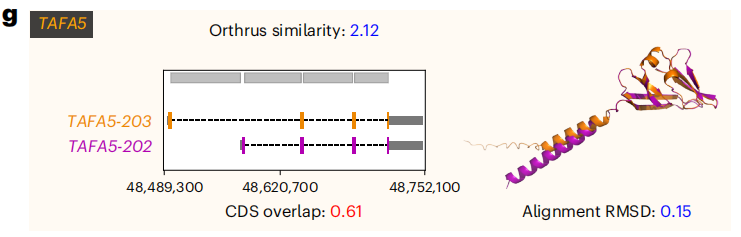
这表明,Orthrus的嵌入空间准确地测量了功能相似性,而不仅仅是序列相似性。它像是一位老练的分子生物学家,能透过序列的差异,直抵功能的本质。
解锁剪接异构体的功能秘密:从“一个基因一个功能”到“一个异构体一个故事”
长期以来,注释单个剪接异构体的功能是RNA生物学的一大挑战。Orthrus证明它或许能解决这个问题。通过对BCL2L1和OAS1这两个著名的功能分化基因进行案例分析,Orthrus没有辜负期望。
在BCL2L1中,Orthrus将促进细胞凋亡的BCL2L1-202和205异构体与其他抑制凋亡的异构体清晰地聚类为两群。在OAS1中,它在潜在空间中将p42和p46这两种具有不同细胞定位和抗病毒功能的异构体正确分离。这证明了Orthrus有能力从单个序列中学习到异构体层面的功能,这对理解神经发育、癌症等疾病至关重要。

意义、局限与未来的挑战
Orthrus的工作,其意义远不止于一个更高效的模型。它揭示了一个更深刻的方向:在AI for Science中,领域知识是最优的“算法”,而进化则是终极的“数据生成器”。
它告诉我们,盲目地扩大参数和堆砌数据可能是缘木求鱼。与其让模型记住所有序列,不如教会它建立功能之间的联系。这种思路将深刻影响未来的生物学基础模型设计:如何更好地利用结构和调控信息?如何训练模型“理解”而非“记忆”?
当然,Orthrus也并非完美。论文讨论部分坦诚地承认,其对比学习目标的潜在代价是可能会抹去对预测特定性质至关重要的信号。例如,在预测某些需要序列细微差别的任务上,强行聚拢异构体可能会丢失信息。此外,模型目前专注于成熟mRNA,尚未纳入非编码RNA和整个pre-mRNA调控的复杂全貌。
但瑕不掩瑜。Orthrus像一束光,照亮了基础模型在生命科学领域可行的、理智的“小而美”的路线。它让那个从30个样本中就能学会预测RNA命运的梦想,变得无比现实。
当我们津津乐道于“暴力美学”在大模型领域的统治力时,Orthrus的故事让我们看到了另一种精彩的博弈。它用700倍的参数效率撬动了同等甚至更优的性能,用生物学直觉而非算力堆砌解决了实际问题。这不禁引发一个更深层次的思考:在追求通用人工智能的道路上,我们是在试图创造一个无所不知的“神”,还是在努力成为一名理解万物的“谦逊”学生? Orthrus用自己的方式给出了它的答案。未来,当其他模型还在疯狂“卷”参数的时候,我们是否应该回头看看,进化这个存在了亿万年的“最伟大教师”,早就为我们标好了所有的重点?欢迎在评论区留下你的见解。

AtomGit 是由开放原子开源基金会联合 CSDN 等生态伙伴共同推出的新一代开源与人工智能协作平台。平台坚持“开放、中立、公益”的理念,把代码托管、模型共享、数据集托管、智能体开发体验和算力服务整合在一起,为开发者提供从开发、训练到部署的一站式体验。
更多推荐
 5
5 0
0- 0



 已为社区贡献16条内容
已为社区贡献16条内容

所有评论(0)