预见基因表达,DNA序列只讲了一半的故事:给AI模型一扇观测染色质“开关”的窗户
论文信息
标题:btag199
预见基因表达,DNA序列只讲了一半的故事:给AI模型一扇观测染色质“开关”的窗户
一句话速览: 大多数预测基因表达的AI模型只看DNA序列,忽略了染色质是否“打开”这个关键开关。一项新研究证明,直接把染色质可及性数据作为输入特征喂给模型,预测精度显著飙升,尤其在那些最难搞定的“高度可变基因”上。更妙的是,这个“加个通道”的策略极其简单,几乎可以移植到任何现有模型上。
当AI只学会了一半的基因调控
如果要预测一个基因是否会被表达,最直接的传统做法是看它的“启动子”DNA序列。就像预测一场演讲是否精彩,先看看演讲稿写得怎么样。
这个逻辑催生了一大批“序列到表达”(Sequence-to-expression)模型。它们用卷积神经网络(CNN)读取DNA上的字母(A、T、C、G),试图从中解码出基因的活动规律。这些模型确实取得了成功,但它们忽略了一个生物学上的常识:就算演讲稿再完美,如果演讲厅的门是锁着的,听众根本进不来。
这个“门”,就是染色质的可及性(Chromatin Accessibility)。DNA在细胞里不是裸着的,它像线圈一样缠绕在组蛋白上。当某段DNA被紧密缠绕时,转录机器无法靠近,基因就无法表达;只有当这段DNA“松开”,变得可及(Accessible),转录才能发生。这个“松开”的状态,可以通过ATAC-seq等实验技术测量出来。
现有的主流模型在干什么?它们要么只盯着DNA序列,要么在多任务学习中“顺便”学一下表观基因组特征。但鲜有人尝试一个最直接、最优雅的操作:把染色质可及性当成一个额外的输入通道,就像给AI的图像模型增加一个“深度”通道一样。
Lapohos等人在(Bioinformatics, 2022)发表的研究,就是为了填上这个显而易见的空白。他们提出的假设非常朴素:与其让AI模型在茫茫DNA序列里大海捞针,不如直接告诉它“哪些区域的门是打开的”。
一个极其简单的“加通道”操作
这个团队所做的,在技术上令人难以置信地简洁。
想象一个标准的卷积神经网络。它的输入通常是一个4通道的矩阵,分别代表DNA的4种碱基(A、T、C、G)。现在,作者在这个4通道旁边,加上了第5个通道——一条浮点数值的曲线,代表该基因启动子区域在特定细胞类型中的ATAC-seq信号强度。
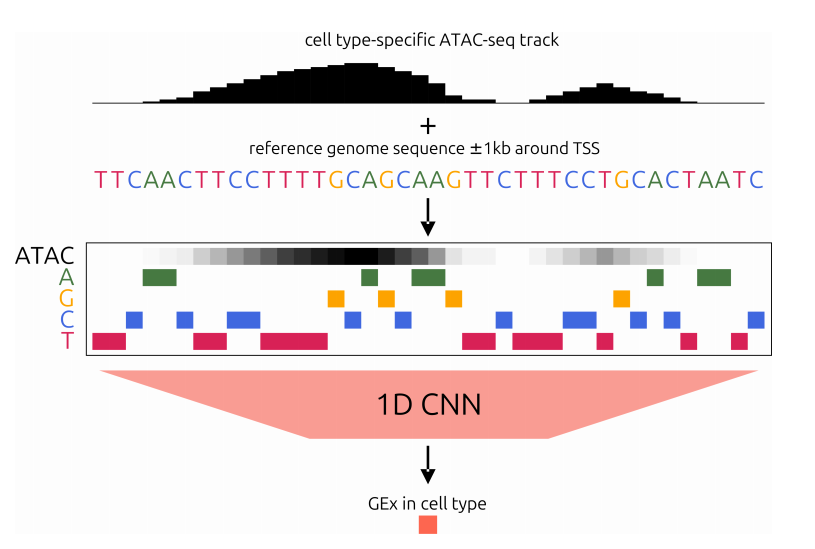
就这么简单。不需要复杂的双模态编码器,不需要Transformer,甚至不需要改变模型的骨架。他们选用了轻量级的CNN架构(基于Xpresso),将输入序列限定在转录起始位点(TSS)上下游各1kb的范围内。
这个设计背后的直觉是:一旦AI知道了哪些DNA片段是“打开”的,它就能更聚焦地学习这些片段的序列特征,而不是被大量封闭的、不相关的序列噪音所干扰。
为了验证这个想法的有效性,研究团队设计了严密的消融实验(Ablation study)。他们训练了三组模型:
-
DNA-only:只看DNA序列(4通道)。
-
ATAC-only:只看染色质可及性数据(1通道)。
-
DNA+ATAC:两者都看(5通道)。
他们使用了来自10x Genomics的三个人类多组学数据集(外周血单核细胞、大脑、空肠),涵盖了12种不同的细胞类型,并用嵌套交叉验证确保结果的稳健性。
数据不会说谎:精度全面提升
实验结果是压倒性的。
在PBMC(外周血单核细胞)数据集上,只看序列的DNA-only模型平均皮尔逊相关系数为0.366。而DNA+ATAC模型一举将这个数字提升到了0.534。
更值得注意的发现来自以下的几个关键实验:
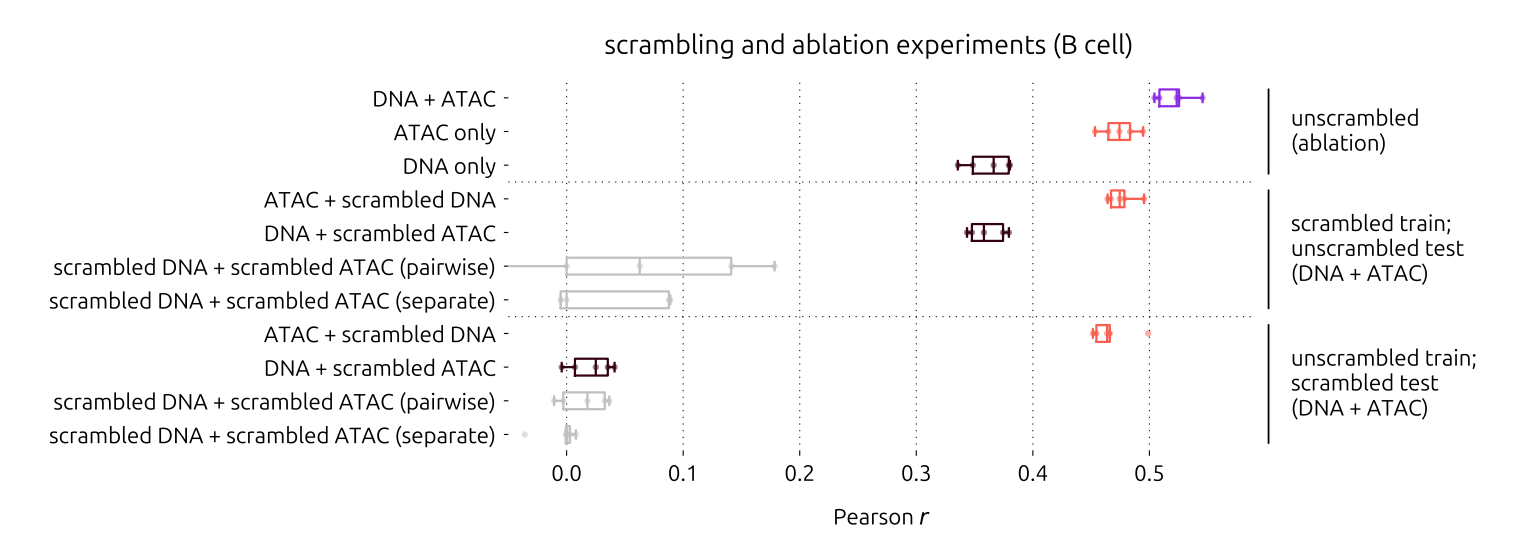
1. 疯狂打乱输入数据
为了证明不是模型在“偷懒”或正好撞上数据中的巧合,研究团队做了“打乱实验”(Scrambling experiment)。他们分别打乱DNA序列和ATAC轨迹与基因表达的对应关系。
结果如何?打乱DNA时,模型的预测能力暴跌至和ATAC-only模型一样;打乱ATAC时,预测能力则和DNA-only模型持平。当两者都被打乱时,模型基本丧失了预测能力。这有力地证明了:DNA+ATAC模型确实是从这两个不同来源的输入中提取了互补信息,而不是单纯依赖一个更强信号。
2. 挑战“最难预测”的基因
基因表达预测中最棘手的任务是什么?是预测那些表达水平在细胞间剧烈波动的“高度可变基因”(Highly Variable Genes)。这些基因通常不是维持细胞基本功能的“管家基因”,而是决定细胞身份和状态的关键调节因子。
研究发现,所有模型在高度可变基因上都表现得更差,但DNA+ATAC模型相比DNA-only模型的相对提升幅度最大。这表明,引入染色质可及性信息,实实在在地帮助模型捕捉到了那些只在一部分细胞中打开的“开关”,而不仅仅是靠猜平均表达水平。
3. 模型学会了看“地图”而不是“蛮力记忆”
AI模型的“黑箱”问题一直为人诟病。为了解开这个黑箱,研究者使用SHAP(Shapley Additive Explanations)工具计算了每个输入位置对模型预测的贡献度(Attribution Score)。
在最关键的发现中,他们观察到:在DNA+ATAC模型中,DNA输入通道的SHAP分数与ATAC输入轨迹之间的相关性显著提高了。这意味着,一旦模型知道了哪里染色质是开放的,它在解读DNA序列时就会在这些开放区域投入更多的“注意力”。模型不再是一项一项地阅读所有碱基,而是学会了聚焦在那些“门开着”的窗口。
背后的生物学:发现了真正的“操盘手”
这个模型带来的不仅仅是数字上的提升,它还揭示了一些生物学规律。
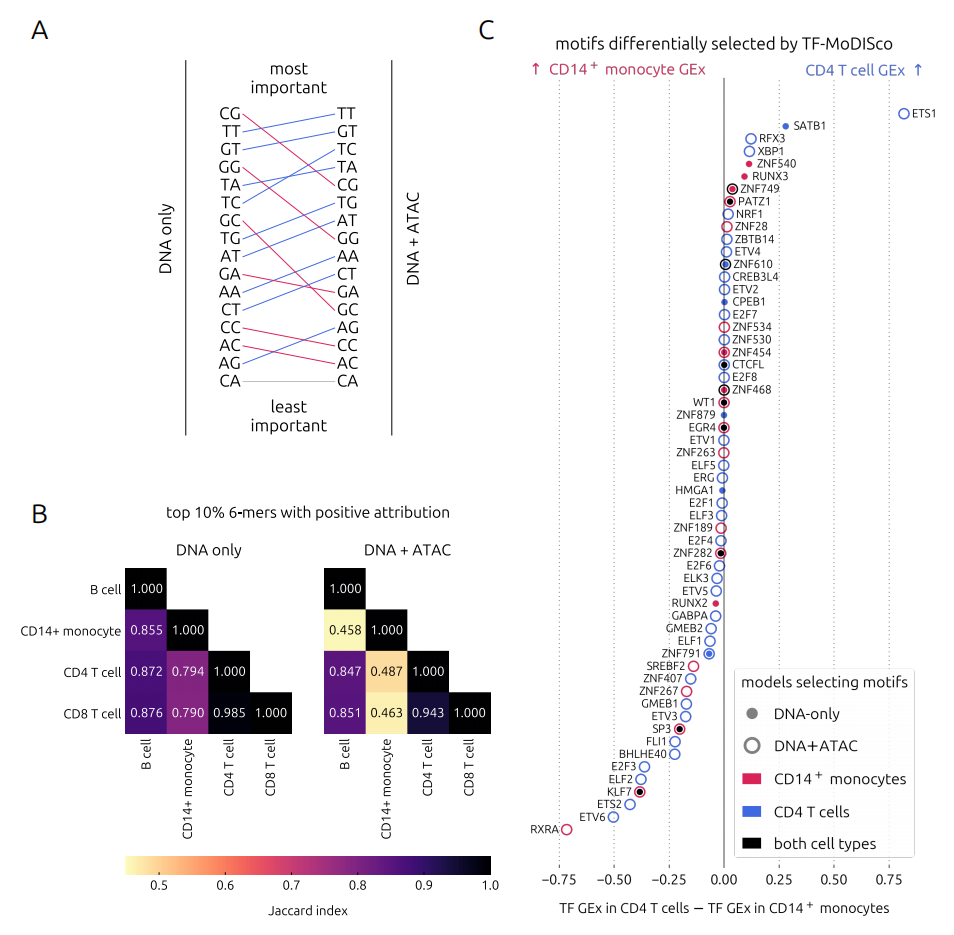
通过对k-mer(长度为k的DNA短序列)的归因分析,研究者发现:DNA+ATAC模型对CpG二核苷酸的依赖大大降低了。 CpG岛在启动子中非常常见,DNA-only模型可能会过度依赖这个简单的特征。而有了ATAC信息的加入,模型被迫去寻找更重要的、有功能的序列模式。
更精彩的是细胞类型特异性motif(基序)的发现。通过TF-MoDISco工具,研究者对比了CD14+单核细胞和CD4+ T细胞的模型。结果发现:
-
在CD14+单核细胞的DNA+ATAC模型中,特异性地发现了RXRA基序。这完全合理,因为RXRα是单核细胞发育维持的关键转录因子。
-
在CD4+ T细胞的DNA+ATAC模型中,则特异性地发现了ETS1基序,ETS1正是对T细胞功能至关重要的因子。
DNA-only模型完全错过了这些清晰的特异性信号。更令人兴奋的是,DNA+ATAC模型还能捕捉到GATA2这种“先驱因子”(Pioneer Factor)的基序。先驱因子的一大特点就是能结合封闭的染色质。这个发现说明,同时输入序列和可及性数据,模型反而学会了区分“通过序列结合”和“通过开放状态结合”这两种不同的调控模式。
提升性能的“组合拳”:预训练
除了直接拼接,研究团队还探索了一种更精巧的策略:先在大规模数据上预训练一个DNA-only模型,再用少量数据加上ATAC通道进行微调。
这个策略听起来像是“我先把演讲稿背熟了,再去看会场地图”。结果非常理想:经过预训练的微调模型,在预测性能上显著优于直接从随机权重开始训练的DNA+ATAC模型。
这种“两步走”策略的实用性很强,因为在现实世界,大规模的DNA序列数据和细胞类型特异的ATAC数据常常不是同时获取的。先利用海量的普通数据训练基础模型,再针对特定细胞类型进行精细调校,显然是一种更具操作性的方案。
这不是终点,而是起点
这项工作的价值在于它的简洁性和可移植性。正如作者强调的,“这种策略可以轻易地适配到任何序列到表达模型上”。
当然,任何研究都有其边界。该模型目前在预测变异效应(eQTL)上表现不佳,这主要是因为模型预测的是“基因表达的概率”而非“转录本的丰度”。此外,2kb的输入序列长度虽然保证了计算效率,但也忽略了对远距离增强子(Enhancer)的考虑。不过,作者进行的13.5kb长序列实验显示,增加输入长度后,DNA+ATAC模型的优势依然存在。
这篇研究给整个领域提了一个醒:在未来设计预测系统时,不要只盯着DNA序列——别忘了那个“打开”了在哪些地方的窗户。
如果我们可以如此简单地通过引入已知的生物学调控指征(染色质可及性)来提升预测精度和解释性,那么未来是否还会有其他更复杂的调控层级,比如三维基因组结构或翻译后修饰,也被这样“加个通道”地整合进去,从而最终拼凑出一张真正的、全息的基因调控图谱?

AtomGit 是由开放原子开源基金会联合 CSDN 等生态伙伴共同推出的新一代开源与人工智能协作平台。平台坚持“开放、中立、公益”的理念,把代码托管、模型共享、数据集托管、智能体开发体验和算力服务整合在一起,为开发者提供从开发、训练到部署的一站式体验。
更多推荐

 6
6 0
0- 0



 已为社区贡献16条内容
已为社区贡献16条内容

所有评论(0)