滇桐(Craigia yunnanensis)染色体水平基因组--文献精读224
A chromosome-level reference genome of an endangered plant Craigia yunnanensis
濒危植物滇桐(Craigia yunnanensis)染色体水平参考基因组
滇桐(Craigia yunnanensis)为东亚特有濒危物种,具备重要的经济价值与科研价值。目前,由于缺乏参考基因组,严重制约了滇桐遗传变异及保护管理相关研究的开展。为弥补这一研究空白,本研究结合 PacBio HiFi 测序与 Hi-C 染色体挂载技术,组装获得了高质量、染色体水平的滇桐参考基因组。该基因组全长 1618.96 Mb,scaffold N50 长度为 39.39 Mb,其中 98.00% 的序列成功挂载至 41 条染色体;BUSCO 评估显示基因组完整度高达 99.40%。研究共预测得到 58969 个蛋白编码基因,其中 94.09% 的基因完成功能注释。本次滇桐染色体水平基因组的成功组装,有助于深入解析滇桐属植物的适应性进化机制,也为该濒危物种的保护保育与科学化管理提供关键的理论依据与数据支撑。
背景与摘要
滇桐(Craigia yunnanensis W. W. Sm. & W. E. Evans)为锦葵科落叶乔木,是东亚特有珍稀树种。该物种主要分布于中国南方及越南北部的石灰岩山地林区,野生种群呈小面积、片段化分布。因大规模森林砍伐,其自然栖息地已严重退化与破碎化。滇桐目前被列为国家保护物种,并在IUCN 红色名录中被评定为易危(VU)等级,具有极高的保护价值。
濒危物种保护的核心目标是保护物种的遗传变异,包括遗传多样性与种群结构,这是制定高效保护策略的基础。已有研究采用分子标记技术对滇桐种群结构开展分析。随着高通量测序技术的发展,物种遗传变异评估进入全新阶段。全基因组测序不仅能提供更可靠的系统发育关系、遗传多样性与种群结构数据,还可为解析物种起源与分化、历史有效种群规模及适应性进化提供关键依据。
然而,目前缺乏高质量滇桐参考基因组,严重制约其遗传变异与保护管理研究。若无染色体水平组装,核基因、结构变异等关键基因组特征无法解析,进而降低系统发育分析精度,难以支撑科学保护策略制定。近期已有研究完成滇桐叶绿体基因组测序,用于分析其系统发育位置。但叶绿体基因组仅占物种总遗传信息的一小部分,绝大部分遗传信息位于核基因组中。因此,构建高质量、染色体水平的滇桐基因组,对全面解析滇桐属系统发育关系、推动濒危滇桐的保护研究至关重要。
本研究整合PacBio HiFi 测序(51.44 Gb)、Illumina 短读长测序(94.54 Gb)与 Hi-C 测序(176.00 Gb) 技术,成功组装获得高质量、染色体水平的滇桐基因组。基因组总长度约 1618.96 Mb,contig N50 达 34.28 Mb;其中 1586.60 Mb(98.00%)序列挂载至 41 条假染色体,scaffold N50 为 39.39 Mb。重复序列占基因组 71.58%,以长末端重复序列(LTRs)为主,占比 54.48%。研究共注释得到58969 个蛋白编码基因。该高质量基因组为解析滇桐的适应性进化机制提供了核心遗传资源。
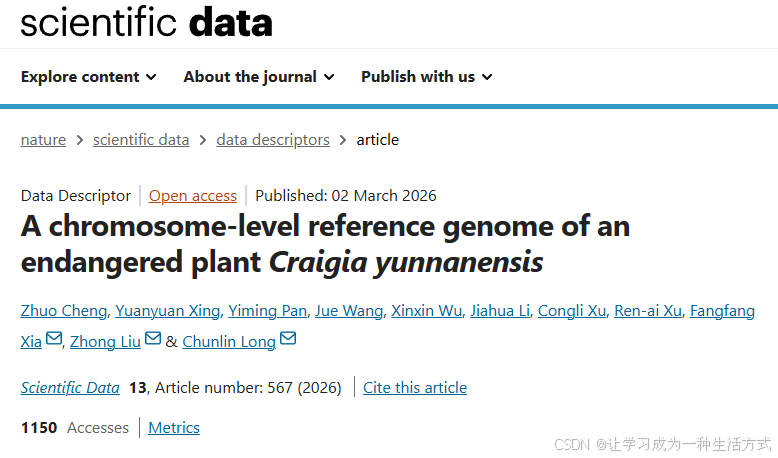
滇桐植株形态与采样位点
(a) 滇桐形态特征;(b) 样本采集地理位置。
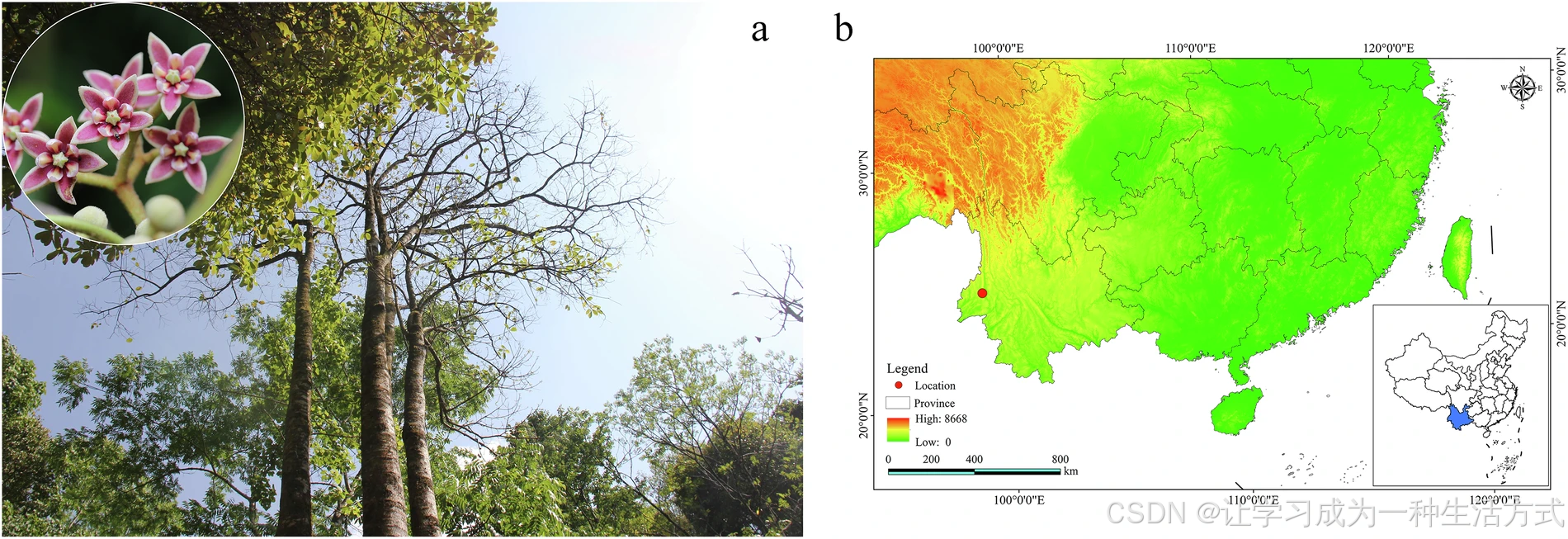
滇桐核型分析与基因组预估分析
(a) 滇桐染色体核型分析;(b) 基于 21-mer 分布的基因组大小预估;(c) 利用 Smudgeplot 工具进行倍性推断。
Illumina 测序与基因组预估分析
采用 NovaSeq 平台构建 150 bp 双端测序文库并完成 Illumina 测序。转录组测序(RNA-seq)方面,使用NEBNext® Ultra™ II 定向 RNA 建库试剂盒提取所有组织样本的总 RNA,同样在 Illumina NovaSeq 平台获得 150 bp 双端读长数据。为预估滇桐基因组大小,委托北京安诺优达基因科技有限公司构建 150 bp 插入片段 DNA 文库并开展二代测序。过滤低质量读长后,共获得 94.54 Gb 有效清洁数据用于基因组调研分析。使用 Jellyfish v2.2.9 统计 21-mer 频率分布,结合 GenomeScope v2.0 估算基因组大小与杂合率。k-mer 分析结果显示,滇桐预估基因组大小为 1619.95 Mb,基因组杂合度为 0.78%(图 2b)。利用 FastK v1.1 分析 k-mer 分布,再通过 Smudgeplots v0.5.0 评估倍性;依据 k-mer 分型特征,证实滇桐为二倍体物种(图 2c)。
HiFi 测序与基因组草图组装
参照太平洋生物科学公司(美国加州)官方实验方案,采用 SMRTbell Express Template Prep Kit 2.0 构建 SMRTbell 环状文库,于 PacBio Sequel II 平台完成 HiFi 长读长测序。过滤低质量序列后,获得 51.44 Gb 高质量清洁数据用于基因组组装。使用 Hifiasm v0.15.4 软件,设置参数 -l 3 完成 HiFi 读长无参从头组装。最终基因组草图总长 1.61 Gb,包含 474 条 contig,contig N50 长度达 34.28 Mb(表 1)。该组装总长与 k-mer 预估的 1619.95 Mb 基因组大小高度吻合。
|
Assembly feature |
Statistics |
|
Genome size (bp) |
1,618,965,816 |
|
Genome size in chromosomes (bp) |
1,586,601,653 |
|
Pseudochromosomes |
41 |
|
GC content (%) |
34.79 |
|
Maximum contig length (bp) |
58,672,620 |
|
Contig N50 length (bp) |
34,285,331 |
|
Contig N90 length (bp) |
15,766,680 |
|
Scaffold N50 length (bp) |
39,392,363 |
|
Scaffold N90 length (bp) |
29,186,167 |
|
BUSCO completeness (%) |
99.4 |
|
LTR Assembly Index (LAI) |
11.61 |
|
QV |
54.22 |
|
Number of protein-coding genes |
58,969 |
|
Percentage of repeat sequences (%) |
71.58 |
|
LTR content (%) |
54.48 |
|
Gypsy content (%) |
7.63 |
|
Copia content (%) |
46.86 |
Hi-C 测序与染色体水平组装
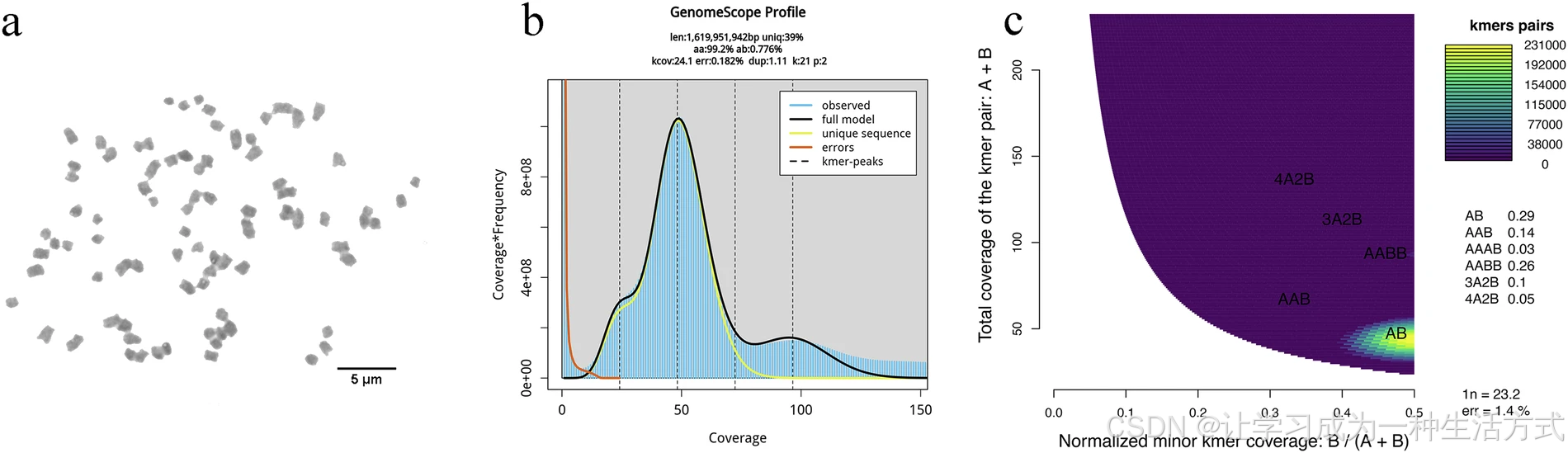
Hi-C 测序实验中,首先利用甲醛对基因组 DNA 进行交联固定,随后完成核酸提取与文库构建,采用双端测序策略于 Illumina NovaSeq 平台(美国加州)上机测序。全部 Hi-C 实验由北京安诺优达基因科技有限公司完成。为实现染色体水平基因组组装,首先通过 Juicer v1.6.1 将 Hi-C 有效测序读长比对至滇桐 contig 组装序列;再利用 3D-DNA v20100812 完成重叠群的初步定位与方向锚定。为降低组装错误,使用 Juicerbox v2.20.00 对 3D-DNA 初步结果进行纠错优化,修正错误连接并优化 contig 聚类。校正后的序列经由 3D-DNA 挂载组装为假染色体,并通过 HiCExplorer v3.7.4 绘制 Hi-C 互作热图。最终,借助 Hi-C 数据共将 1586.60 Mb 组装序列(占比 98.00%)成功锚定到 41 条假染色体上,染色体长度区间为 23.53 Mb–58.67 Mb(表 1、图 3a,b)。
滇桐 Hi-C 互作关系及基因组组装特征
(a) 滇桐 41 条假染色体的 Hi-C 染色质互作热图;(b) 基因组组装长度与实际染色体物理长度的相关性;(c) 滇桐全基因组特征圈图。Circos 圈图由外至内依次为:染色体水平假染色体、基因共线性(曲线连线)、GC 含量、基因密度、转座子密度、长末端重复序列密度。
基因组注释
结合同源比对预测与从头预测两种策略,对滇桐基因组的转座元件及功能基因进行鉴定与注释。转座元件鉴定:使用 RepeatModeler v2.0.6,添加 -LTRStruct 参数构建物种特异性从头重复序列文库;再整合 Repbase(v20181026)与 Dfam(v3.7)数据库的同源重复序列库,通过 RepeatMasker v4.0.9 完成全基因组重复序列筛查。整合两种预测结果统计显示:滇桐基因组重复序列总长约 1135.63 Mb,占基因组全长 71.58%(表 1、表 2)。各类重复元件中,** 长末端重复反转座子(LTR)** 占比最高,达 54.48%;DNA 转座子次之,占比 13.82%。
|
Classification |
De novo + Repbase |
TE Proteins |
Combined TEs |
|||
|
Length (bp) |
Ratio (%) |
Length (bp) |
Ratio (%) |
Length (bp) |
Ratio (%) |
|
|
DNA |
219,156,995 |
13.81 |
2,970,770 |
0.19 |
219,208,328 |
13.82 |
|
LINE |
20,385,460 |
1.28 |
192,867 |
0.01 |
20,405,432 |
1.29 |
|
SINE |
3,452,001 |
0.22 |
0 |
0.00 |
3,446,316 |
0.22 |
|
LTR |
863,221,725 |
54.41 |
244,436,832 |
15.41 |
864,432,269 |
54.48 |
|
LTR-Gypsy |
120,795,571 |
7.61 |
44,916,713 |
2.83 |
121,015,239 |
7.63 |
|
LTR-Copia |
742,426,154 |
46.79 |
199,520,119 |
12.58 |
743,417,030 |
46.86 |
|
Satellites |
833,129 |
0.05 |
0 |
0.00 |
833,129 |
0.05 |
|
Unclassified |
27,308,968 |
1.72 |
176 |
0.00 |
27,307,735 |
1.72 |
|
Total |
1,134,358,278.00 |
71.50 |
247,600,645.00 |
15.61 |
1,135,633,209.00 |
71.58 |
蛋白编码基因注释整合了同源比对证据、RNA-seq 数据与从头预测三种方法。基于同源序列的基因模型使用 GeMoMa v1.9 构建,比对物种包括拟南芥(Arabidopsis thaliana)、木棉(Bombax ceiba)、可可(Theobroma cacao)和榴莲(Durio zibethinus)的蛋白序列。利用这些训练集训练了两种从头基因预测软件:Augustus v3.5.0 与 GENEMARK-ES v4.72。同时,使用 Hisat2 v2.2.1、StringTie v2.2.3 和 TransDecoder v5.7.1 完成转录组无参组装。将训练好的预测模型、同源证据与转录组组装结果通过 EvidenceModeler v2.1.0 流程整合,确保准确识别转座元件并全面注释滇桐基因组的蛋白编码基因。
本研究结合从头预测、同源比对和 RNA-seq三种策略对滇桐进行基因鉴定与注释,最终共预测得到 58,969 个蛋白编码基因(表 3)。其中,55,485 个基因(94.09%) 可在至少一个数据库中获得功能注释,包括:NCBI 非冗余蛋白数据库(NR)、UniProt、Pfam、真核生物同源蛋白群(KOG)、基因本体(GO)、京都基因与基因组百科全书(KEGG)(图 4a)。
|
Methods |
Software/Species |
Number |
Average gene length (bp) |
Average CDS length (bp) |
Average exons per gene |
Average exon length (bp) |
Average intron length (bp) |
|
De novo |
Augustus |
45,851 |
5,051.27 |
1,518.16 |
6.30 |
285.08 |
536.40 |
|
GeneMark-ES |
81,715 |
2,946.05 |
1,074.08 |
5.20 |
206.61 |
447.15 |
|
|
Homolog |
Bombax ceiba |
54,423 |
3,186.33 |
1,231.38 |
5.20 |
235.01 |
461.11 |
|
Durio zibethinus |
53,019 |
3,949.12 |
1,340.02 |
6.00 |
224.16 |
524.13 |
|
|
Arabidopsis thaliana |
48,473 |
3,354.34 |
1,282.40 |
5.70 |
225.98 |
443.21 |
|
|
Theobroma cacao |
56,067 |
3,357.53 |
1,310.07 |
5.60 |
235.63 |
449.02 |
|
|
RNA-seq |
TransDecoder |
38,021 |
4,870.73 |
1,286.29 |
5.70 |
313.67 |
446.58 |
|
EVM |
58,969 |
3,557.09 |
1,298.63 |
5.70 |
227.30 |
479.18 |
|
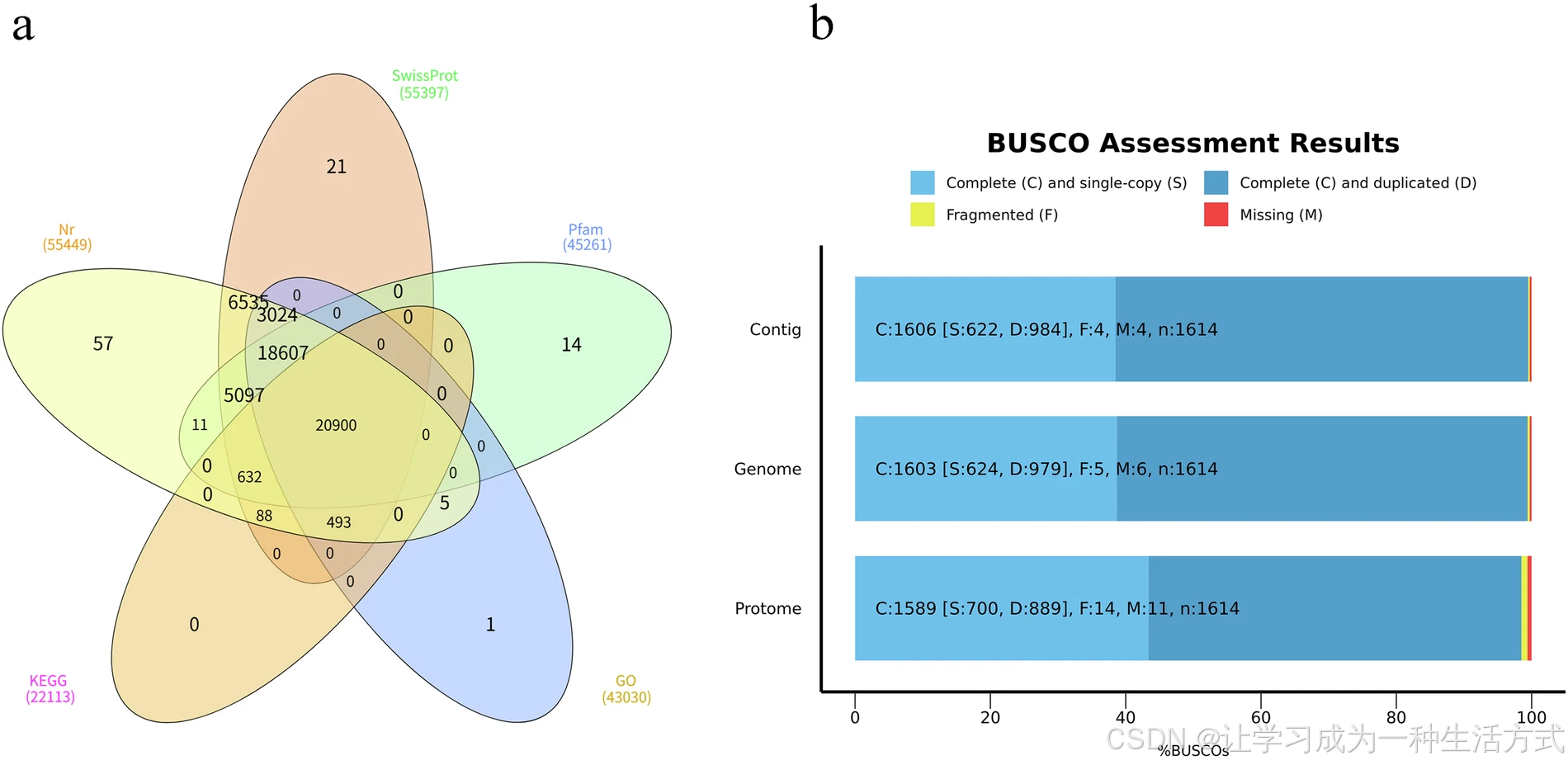
滇桐基因组组装与注释质量评估
(a) 滇桐蛋白质组注释结果统计(b) 滇桐基因组 contig 水平、染色体水平及蛋白质组的 BUSCO 评估
使用 Infernal v1.1.5 比对 Rfam 数据库鉴定非编码 RNA,包括 miRNA、rRNA 和 snRNA;使用 tRNAscan-SE v2.0.9 预测 tRNA 基因。除蛋白编码基因外,本研究共鉴定出:372 个 miRNA、999 个 tRNA、359 个 rRNA 和 6,330 个 snRNA(表 4)。
|
Type |
Class |
Number |
Total length(bp) |
Average length(bp) |
|
miRNA |
miRNA |
372 |
46,444 |
124.85 |
|
tRNA |
tRNA |
999 |
72,074 |
72.15 |
|
rRNA |
28s_rRNA |
11 |
65,101 |
5,918.27 |
|
18s_rRNA |
10 |
22,980 |
2,298.00 |
|
|
8s_rRNA |
338 |
38,831 |
114.88 |
|
|
snRNA |
snRNA |
3,165 |
367,896 |
116.24 |
|
CD-box |
2,099 |
219,510 |
104.58 |
|
|
HACA-box |
122 |
15,608 |
127.93 |
|
|
splicing |
944 |
132,778 |
140.65 |
数据记录
本研究产生的所有原始测序数据已提交至美国国家生物技术信息中心(NCBI)的序列读取存档(SRA)数据库,对应的生物项目编号为 PRJNA1327616。HiFi、Hi-C、Illumina 及 RNA-seq 测序数据集的存档编号分别为:SRR3538602434、SRR3621621835、SRR3537130836、SRR3636973337。
基因组组装序列已提交至 GenBank,登录号为 GCA_054051545.1。基因组组装序列及其注释文件可通过 Figshare 公开获取,链接如下:https://doi.org/10.6084/m9.figshare.3007572739
技术验证
滇桐体细胞为二倍体,包含 41 对染色体,其中:
- 中部着丝粒染色体:16 条
- 近中部着丝粒短染色体:44 条
- 近端着丝粒染色体:22 条
为验证组装质量,我们将体细胞中 41 对染色体的物理长度与组装长度进行比较。结果显示二者高度一致,相关系数为 0.99,p 值为 7.55e-35,表明存在极强的正相关关系(图 3b)。
对滇桐基因组组装的连续性、完整性与准确性进行了综合评估。为验证组装准确性,分别使用 HiFi 与 Illumina 测序读段进行比对:
- HiFi 读段使用 Minimap2 v2.28 比对
- Illumina 读段使用 BWA v0.7.18 比对
- 比对率使用 Samtools v1.17 计算
组装结果显示:
- NGS(Illumina)读段比对覆盖率:99.64%
- PacBio HiFi 读段比对覆盖率:99.91%(表 5)
所有测序读段均能有效比对到滇桐基因组,进一步证实组装的准确性与完整性。
使用 BUSCO v5.4.3 结合 embryophyta_odb10 数据库评估完整性,结果显示 99.4% 的核心基因完整,仅 0.3% 缺失(图 4b)。此外,使用 Merqury v1.3 在 k-mer=20 条件下评估组装准确性,得到平均质量值 QV = 54.22。
以上结果共同表明:滇桐基因组组装具有极高的连续性、完整性与准确性。
|
Reads Type |
Samples |
Clean reads |
Q20 Rate |
Q30 Rate |
Properly mapped |
|
Illumina pair-end reads for genome |
short reads |
623590590 |
97.90% |
93.89% |
621378799 (99.64%) |
|
Pacbio HiFi reads for genome |
HiFi reads |
2525642 |
95.15% |
88.82% |
2523387 (99.91%) |
与 Zhang 等人组装中报道的 57,219 个基因相比,本研究共鉴定出 58,969 个蛋白编码基因。采用 BUSCO v5.4.3 软件结合embryophyta_odb10数据库进行评估,结果显示98.5% 的保守直系同源基因可被完整检出,其中 43.4% 为单拷贝基因,55.1% 为重复基因(图 4b)。
此外,共有 55,485 个基因在 NR、SwissProt、KEGG、KOG、GO 和 Pfam 共 6 个数据库中成功获得功能注释(图 4a)。与近期发表的滇桐单倍体基因组相比,本研究组装质量表现相当,contig N50 约 34 Mb,scaffold N50 约 40 Mb(表 6)。
|
Assembly version |
Species |
Genomesize (Mb) |
GC content (%) |
Contig N50 (Mb) |
Scaffold N50 (Mb) |
BUSCO (genome) |
BUSCO (protome) |
|
GCA_051046955.1 |
Craigia yunnanensis (2n = 4x = 164, neoautotetraploid) |
1531.8 |
34.3 |
34.5 |
41.0 |
C:99.4%[S:41.8%,D:57.6%],F:0.3%,M:0.3%,n:1614 |
C:97.6%[S:50.3%,D:47.3%],F:1.2%,M:1.2%,n:1614 |
|
This study |
Craigia yunnanensis (2n = 2x = 82, diploid) |
1586.6 |
34.79 |
34.3 |
39.4 |
C:99.4%[S:38.7%,D:60.7%],F:0.3%,M:0.3%,n:1614 |
C:98.5%[S:43.4%,D:55.1%],F:0.9%,M:0.6%,n:1614 |
本研究采用 BLASTP 筛选二倍体基因组与同源四倍体单倍型基因组种间及种内同源基因,并基于同源基因,通过 JCVI 软件绘制共线性区块。利用 WGDI 计算旁系同源基因对与直系同源基因对的同义替换率(Ks),结合 JCVI 进一步绘制共线性深度比值与共线性点阵图。
结果显示,两组组装基因组的旁系同源基因对 Ks 峰值均约为 1.5(图 5a、5b)。值得注意的是,直系同源基因对的 Ks 值同样接近 1.5(图 5c),两组基因组间无显著差异。该结果证实,同为滇桐物种的二倍体基因组与同源四倍体单倍型基因组具有高度的基因组同源性。
此外,两组基因组间共线性深度比为 1:1(图 5d),与二者全部 41 条染色体呈现严格一对一的染色体共线性特征相一致(图 5e)。综上,共线性分析充分证实:同源四倍体单倍型基因组与二倍体参考基因组高度吻合。
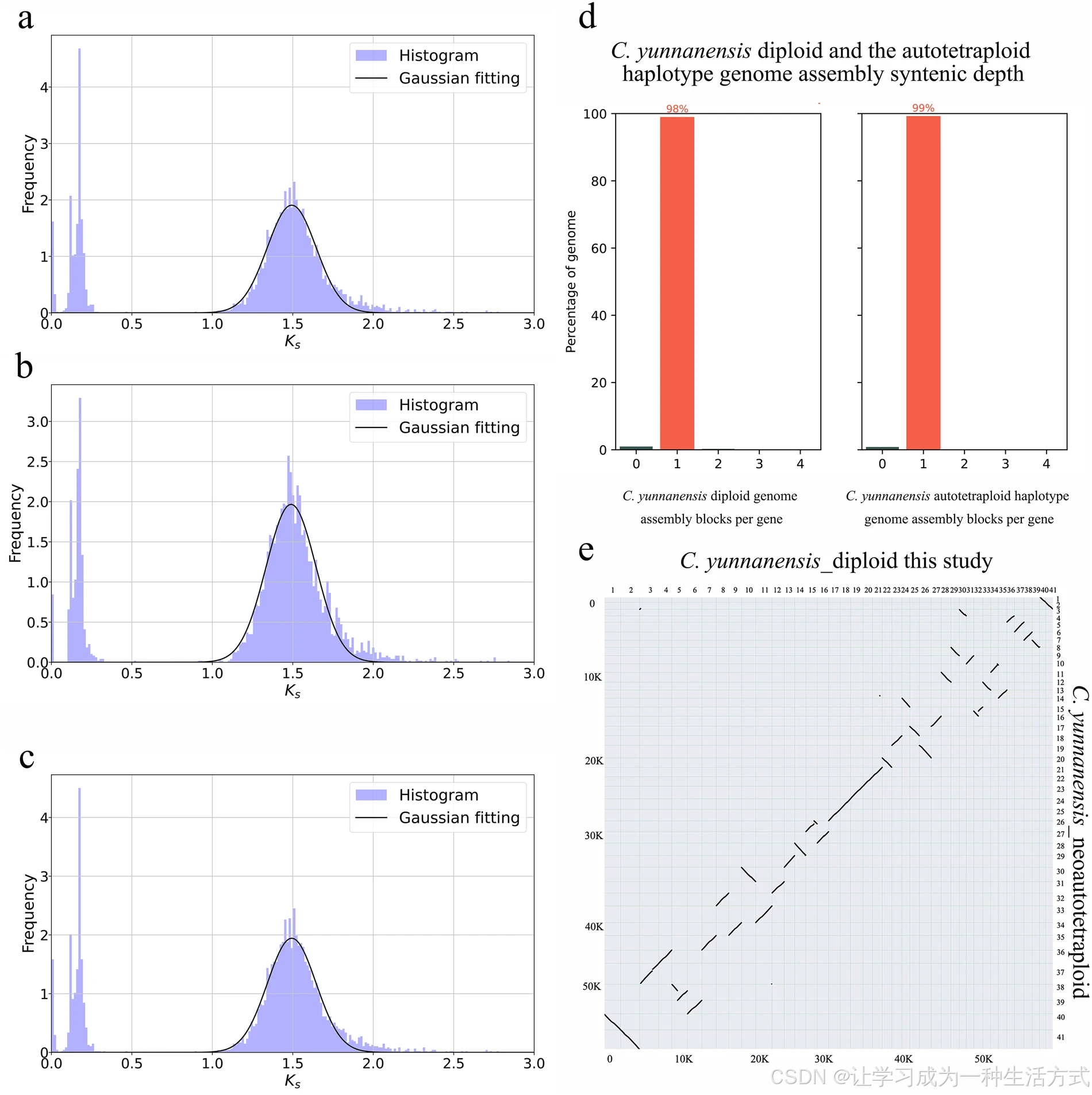
滇桐新生同源四倍体与滇桐二倍体(本研究)的比较分析。(a) 二倍体基因组组装中旁系同源基因对的 Ks 峰值;(b) 同源四倍体单倍型基因组组装中旁系同源基因对的 Ks 峰值;(c) 二倍体与同源四倍体单倍型基因组间直系同源基因对的 Ks 峰值;(d) 二倍体与同源四倍体单倍型基因组的共线性深度比值;(e) 二倍体与同源四倍体单倍型基因组比对分析的共线性点阵图。

AtomGit 是由开放原子开源基金会联合 CSDN 等生态伙伴共同推出的新一代开源与人工智能协作平台。平台坚持“开放、中立、公益”的理念,把代码托管、模型共享、数据集托管、智能体开发体验和算力服务整合在一起,为开发者提供从开发、训练到部署的一站式体验。
更多推荐
 4
4 0
0- 0



 已为社区贡献6条内容
已为社区贡献6条内容

所有评论(0)