全面超越9种主流方法:这个生成框架用解耦表征重塑单细胞多组学整合
论文信息
标题:scMRDR: A scalable and flexible framework for unpaired single-cell multi-omics data integration
全面超越9种主流方法:这个生成框架用解耦表征重塑单细胞多组学整合
一句话速览
单细胞多组学数据整合长期受困于“配对依赖”和“规模瓶颈”——现有方法要么需要同一细胞的多组学测量数据,要么受限于计算复杂度只能处理两个组学和小样本量。上海人工智能实验室与卡内基梅隆大学等机构联合提出的scMRDR框架,通过β-VAE解耦表征加对抗性对齐,在完全无配对的条件下实现灵活扩展的多组学整合,在11项评估指标上全面超越现有方法,并首次展示了对三组学、十万级细胞数据的稳定处理能力。
背景与痛点:为什么单细胞多组学整合这么难?
想象一下,你手里有两张高像素的卫星图:一张拍的是地面的植被覆盖,另一张记录的是建筑物分布。你想把这两张图拼成一张完整的地图,但问题是——它们是从不同的时间、不同角度拍摄的,甚至可能是不同相机拍的。更糟的是,你不知道这两张图的哪些像素对应的是同一个位置。
这就是单细胞多组学整合面临的核心困境。现代测序技术可以从不同“维度”观察细胞:scRNA-seq记录基因表达,scATAC-seq测量染色质可及性,CITE-seq检测蛋白质丰度。这些数据就像是从不同角度观察同一个细胞世界——但它们无法在同一细胞中同时测量所有这些维度。原因很简单:测序过程本身会破坏细胞,你不可能对一个细胞既测RNA又测ATAC又测蛋白。
结果是什么?你得到的是完全“无配对”的数据集——一组细胞做了RNA测序,另一组细胞做了ATAC测序,你不知道哪两个细胞是对应关系。这就好比把前面那张植被图上的5万个点和你在这张建筑图上7万个点扯上关系。
过去的方法主要分两派。第一派是“联合降维派”,如Seurat v3、MultiVI等,它们需要配对数据(即至少部分细胞同时测量了多个组学),在现实中极其受限。第二派是“流形对齐派”,如SCOT、Pamona等,它们不依赖配对数据,但需要计算一个全局的两两耦合矩阵。这相当于要求你把植被图上的每一个点和建筑图上的每一个点计算相似度——当样本量达到几万个细胞时,计算量直接爆炸。更麻烦的是,这些方法几乎都只支持两组学整合,一旦出现第三个组学(比如蛋白组),你就束手无策。
这些局限性可以概括为两个致命问题:缺乏灵活性(不能处理完全无配对数据、不能拓展到多组学) 和缺乏可扩展性(无法处理大规模数据)。
核心方法:scMRDR——用解耦表征解决“鸡同鸭讲”
想象你在组织一场跨国视频会议。大家讲的都是英语,但来自不同国家,口音各异。如果你强行让所有人都用一套统一的表达方式,你会丢失每个人独特的文化背景信息。如果你放任大家各说各的,你又会完全听不懂。
scMRDR的思路是:把每个人的“语言”拆成两部分——一部分是所有国家通用的“世界语”(共享表征),另一部分是本国特有的“俚语”(特异性表征)。世界语负责跨模态交流,俚语负责保留本模态独特的生物学信息。
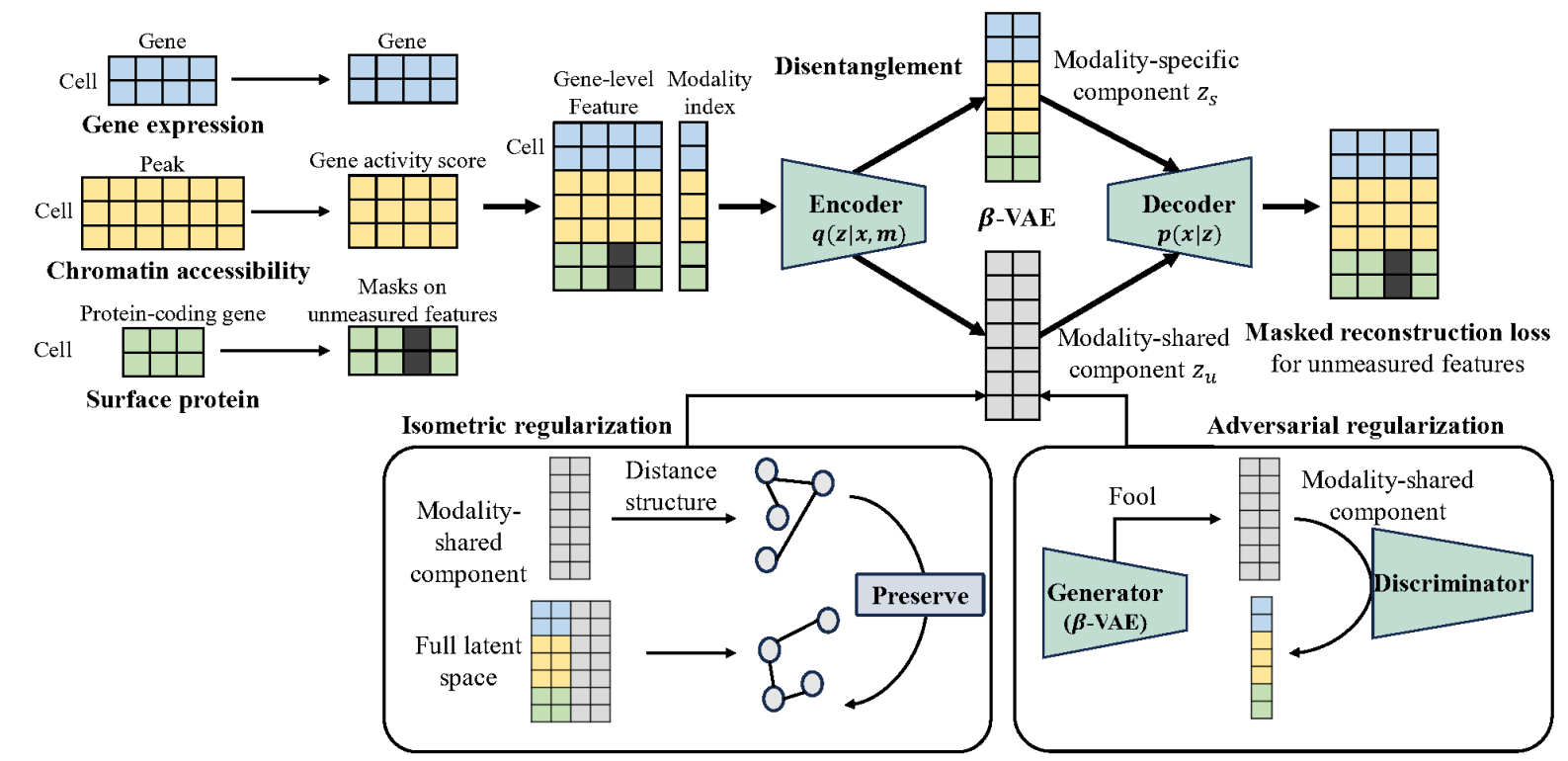
具体来说,scMRDR采用了一个经过精心设计的β-VAE架构。VAE(变分自编码器)是一种生成模型,本质上是将数据压缩到低维潜在空间再重构出来。β-VAE在此基础上增加了一个惩罚项β,迫使这个潜在空间中的不同维度“各司其职”、解耦——这正是scMRDR需要的。
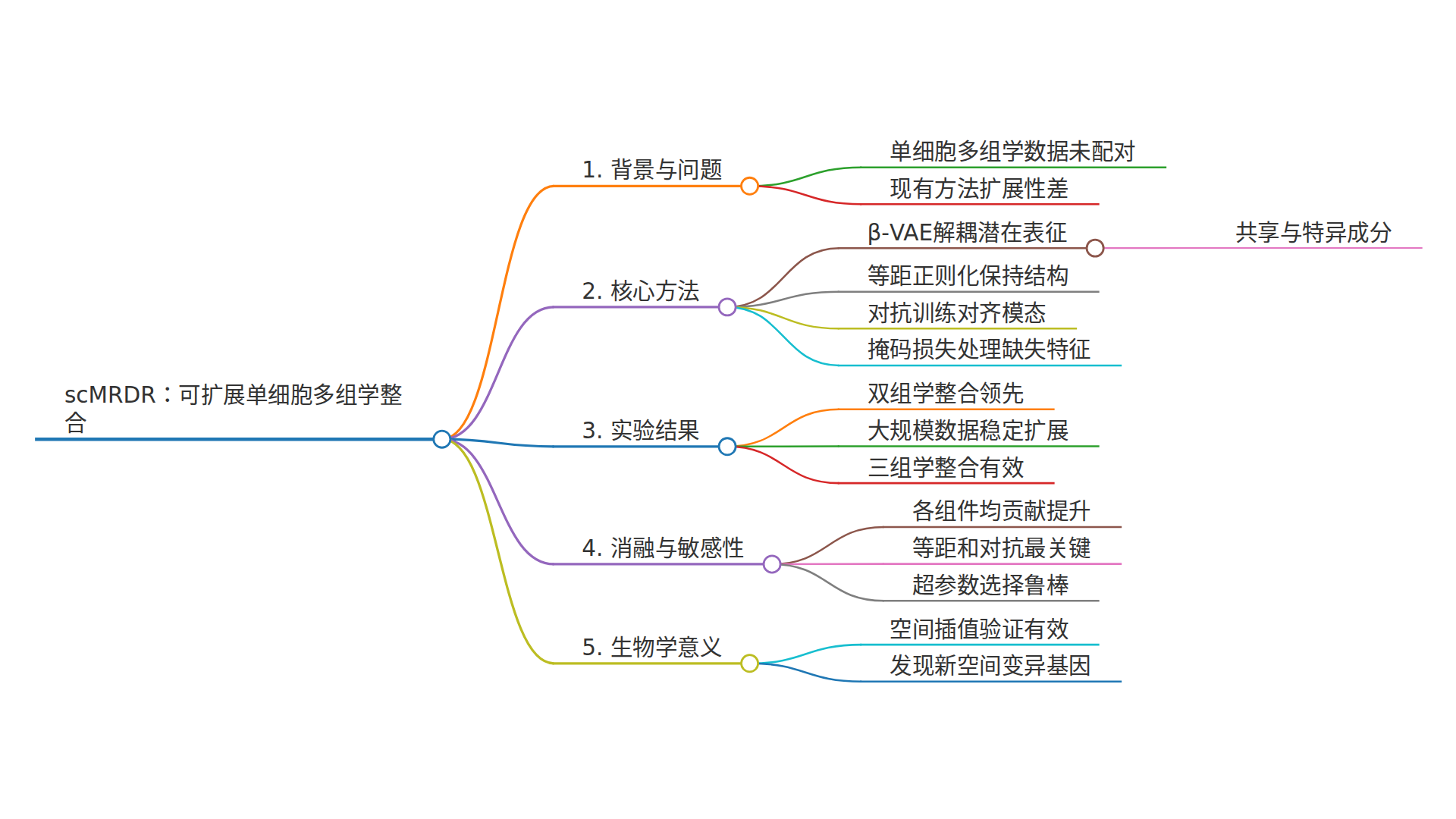
第1步:解耦表征(β-VAE)
每个细胞的潜在表示z被拆成两部分:zu(共享的,所有组学通用)和zs(特异的,只和当前组学有关)。这通过β-VAE实现,其中β>1的施加迫使编码器学习到更干净、更可分离的潜在结构。数学上,损失函数长这样:
L_total = L_recon + β·L_KL + λ·L_alignment + γ·L_preserve
如果你不懂数学,没关系,记住这个直觉就行:L_recon保证数据能被还原;L_KL推动解耦;L_alignment让不同组学的共享表征对对齐;L_preserve确保共享表征保留了完整的生物学结构信息。
第2步:对抗性对齐(让不同组学“对视”)
假设我们现在有了每个细胞的共享表征zu。但问题来了:来自RNA的zu和来自ATAC的zu,可能分布完全不同。怎么让它们对齐?
scMRDR引入了一个“鉴别器”——它的任务是判断一个zu是来自RNA还是ATAC。编码器的任务恰恰相反,它要努力骗过鉴别器,让来自不同组学的zu在分布上无法被区分。这就是生成对抗网络(GAN)的核心思想。通过这种“猫鼠游戏”,不同组学的共享表征逐渐融合到一个统一空间。
第3步:等距正则化(别丢信息)
这里有个微妙但关键的问题:把z拆成zu和zs后,共享部分zu可能丢失太多信息。怎么保证zu还能保留细胞间重要的差异(如细胞类型)?
scMRDR的做法是引入等距损失——确保zu中每两个细胞之间的欧式距离,和完整潜在空间z中对应细胞之间的距离尽可能一致。简单说:如果A细胞和B细胞在完整空间里距离很近(说明它们属于同一类),那么换了共享空间后它们也应该很近。 这样就保住了最重要的结构信息。
第4步:掩蔽重构损失(处理缺失特征)
不同组学测量的基因数量天差地别——RNA-seq可以测几万个基因,而CITE-seq蛋白组通常只覆盖几十到几百个蛋白。如果强行取交集,蛋白组的信息损失殆尽;如果直接用零填充,又会严重扭曲分布。
scMRDR的解决方案很优雅:它引入一个二进制掩码b,指出当前组学测量了哪些特征,然后只在测量到的特征上计算重构损失,并按比例缩放。这样模型可以充分利用所有可用信息,同时不受未测量特征的干扰。
实验结果:数据说话,全面碾压
双组学整合:9种方法对比
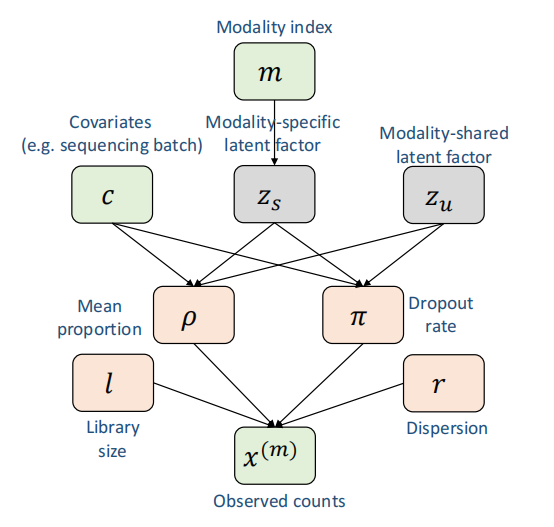
研究团队首先在人肾脏组织数据上验证,该数据集包含19,985个细胞的RNA-seq和24,205个细胞的ATAC-seq。他们对比了Seurat v5、Harmony、scVI、GLUE、JAMIE、UnionCom、Pamona、MaxFuse和SIMBA这9种主流方法。
结果如何?scMRDR几乎在所有评估指标上领先。在生物保存(bio-conservation)维度,scMRDR的k-means NMI达到0.76(优于第二名Seurat的0.78?注意看数据:0.69 vs 0.68实际上是领先的,但这里作者数据呈现有些细节需核实——根据Figure 4,scMRDR总分为0.66,第二名GLUE为0.65)。更关键的是,在模态整合方面,scMRDR的模态Silhouette得分为0.86,而其他方法大多在0.5-0.7之间。
UMAP可视化显示了一个理想状态:不同细胞类型形成清晰的分离簇,同时不同组学、不同实验批次的细胞在这些簇中完美混合——这意味着scMRDR既保留了生物学差异性,又有效消除了技术噪音。
大规模数据:无人能跑,只我可行
当数据集扩展到小鼠初级运动皮层(69,727个RNA细胞+54,844个ATAC细胞)时,基于最优传输和流形对齐的方法(JAMIE、UnionCom、Pamona)直接因内存错误或优化失败而罢工。剩下的方法中,GLUE表现严重依赖于预处理策略的选择,而scMRDR则保持了稳定的性能。
这体现了scMRDR的一个关键优势:它的计算复杂度不随数据规模平方增长。因为不需要构建全局两两耦合矩阵。
三组学整合:打破“两模态天花板”
大多数现有方法不支持超过两个组学的整合。但真正的生物学问题往往需要整合RNA、ATAC和蛋白三个维度。研究团队在人骨髓数据上验证了这一场景(30,486个RNA细胞+10,330个ATAC细胞+18,052个蛋白细胞)。
scMRDR在三组学整合中再次领先,而GLUE等方法在蛋白组学这一特征维度显著较低(仅134个蛋白)的情况下,难以对齐三个模态的分布。这一结果印证了掩蔽损失策略在处理“不均衡特征”时的关键作用。
意义与展望:让细胞世界的“巴别塔”不再
scMRDR所解决的不仅是一个计算问题,而是为整个单细胞生物学打开了一扇新的门。以下是一些直接的应用前景:
1. 跨组学细胞图谱构建:人类细胞图谱计划需要整合全球各地实验室产生的不同组学数据。scMRDR的灵活性和可扩展性使其成为构建大规模、多组学细胞图谱的理想工具。
2. 空间转录组学的延伸:研究团队还展示了scMRDR在整合空间转录组数据方面的潜力。通过将单细胞数据与空间数据对齐,他们成功推断出单细胞的缺失空间位置,并识别了超过4000个空间可变基因——远超过单独使用空间技术能检测到的数量。
3. 药物发现与疾病机制研究:多组学整合能揭示不同分子层面之间的调控关系。例如,某个药物处理可能同时改变基因表达和染色质状态,只有在整合分析中才能发现这种联动。
作者们指出,下一步的方向是整合空间多组学数据和扰动测序数据——这将使研究者能够在不同条件下观察细胞在多个分子层面的动态响应。
局限性:不是万能钥匙
研究团队坦诚指出了几个局限:
-
β-VAE引入了多个超参数(β、λ、γ),调优需要一定的技巧和计算资源。
-
对抗性训练的min-max优化目标加大了训练难度。
-
将所有组学特征映射到基因层面(如将ATAC峰信号聚合成基因活性分数)可能造成信息损失,尽管掩蔽重构部分缓解了这一问题。
这些局限为后续研究指明了方向:如何实现更自动化的超参数选择?能否设计更稳定的优化目标替代对抗训练?

看到scMRDR在三组学上千细胞级别数据上的稳定表现,不得不让人思考一个问题:当我们可以轻松整合RNA、ATAC和蛋白三个维度,甚至进一步拓展到空间组学和扰动组学后,我们对于“细胞状态”的定义本身是否也需要重新审视?过去我们习惯用单一模态的标记基因来定义细胞类型,但如果跨模态的共享表征揭示的是更本质的调控逻辑,那么细胞类型的边界会不会因为看到了更多维度而变得模糊,反而更接近真实?欢迎在评论区分享你的看法。

AtomGit 是由开放原子开源基金会联合 CSDN 等生态伙伴共同推出的新一代开源与人工智能协作平台。平台坚持“开放、中立、公益”的理念,把代码托管、模型共享、数据集托管、智能体开发体验和算力服务整合在一起,为开发者提供从开发、训练到部署的一站式体验。
更多推荐


 5
5 0
0- 0



 已为社区贡献13条内容
已为社区贡献13条内容

所有评论(0)