RF2-PPI论文与代码阅读(人类蛋白质组PPI预测与筛选)
基本信息
论文名:Predicting protein-protein interactions in the human proteome
团队:David Baker实验室
期刊:Science
发表日期:2025.10.23
任务:人类蛋白质组PPI预测
论文链接:Predicting protein-protein interactions in the human proteome | Science
声明:以下的图片与代码全部来自于原论文及其代码仓库
组会上讲的论文,但是讲太快了,许多细节没讲到,其实里面有很多值得学习的内容
论文框架
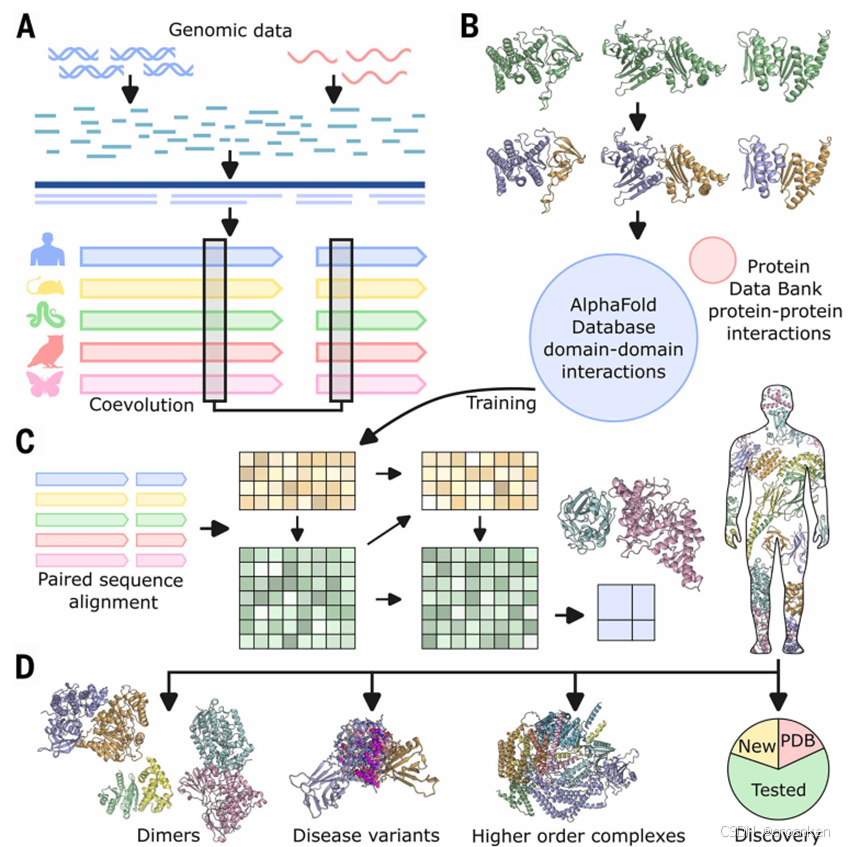
主要流程:
1、omicMSA对人类做共进化分析
2、用AFDB挖掘domain-domain-interaction对
3、RF2-PPI模型预测PPI得分
4、AF2对PPI进行精筛
OmicMSA
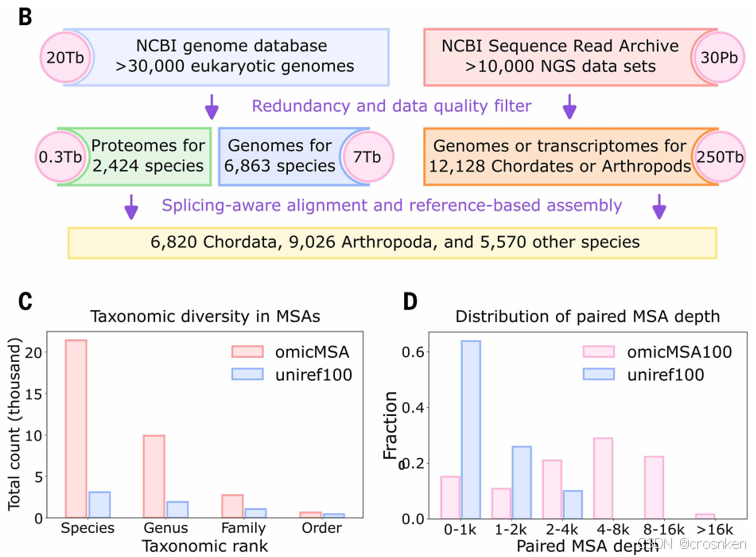
1、蛋白质组构建
把30Pb的原始测序数据SRA通过从头组装,构建了12128个物种的基因组
然后对只有基因组的物种做编码区预测和剪接位点预测(内含子预测),把外显子拼接,构建蛋白质组
一共构建了2w多个物种的蛋白质组
C图是种、属、科、目的覆盖数与uniref100进行的比较
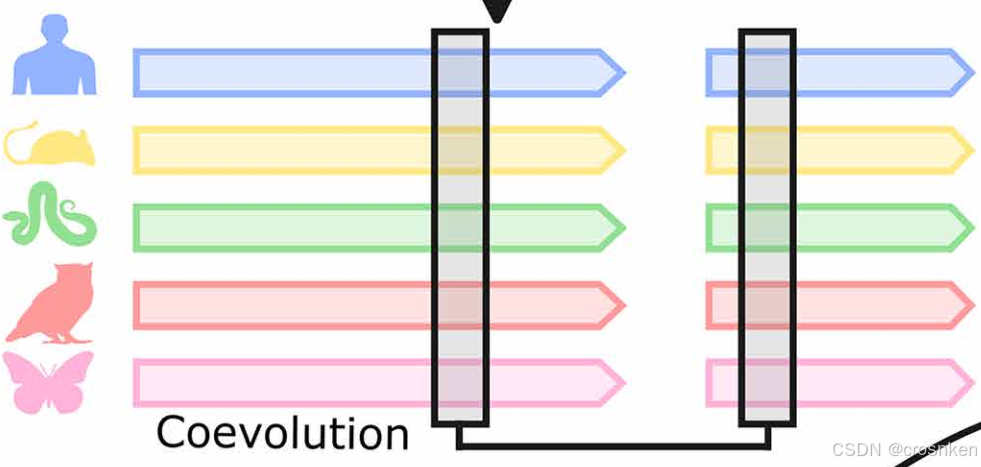
2、直系同源MSA
对人类中的每一个蛋白质p,对每一个物种s都找到MSA得分最高的蛋白质q,再将q放到人类蛋白质组中做MSA,找到得分最高的p’
如果p’=p,则认为该物种中的蛋白质q与人类的蛋白质p直系同源
这样,每个人类蛋白质p都对应了一个直系同源蛋白质q的集合Q(p)={q1,q2,q3,...},和其对应的直系同源物种集合X(p)={x1,x2,x3,...}
3、pair MSA算法(pMSA)
对于蛋白质p1和p2,求它们直系同源物种的交集:X(p1,p2)=X(p1)∩X(p2)
然后在每个交集物种x中,选择出p1,p2各自的直系同源蛋白质(qi,qj),构成集合Q(p1,p2)
把(p1,p2)和Q(p1,p2)排列成字符矩阵的形式,中间用gap连接,如下图
DDI挖掘
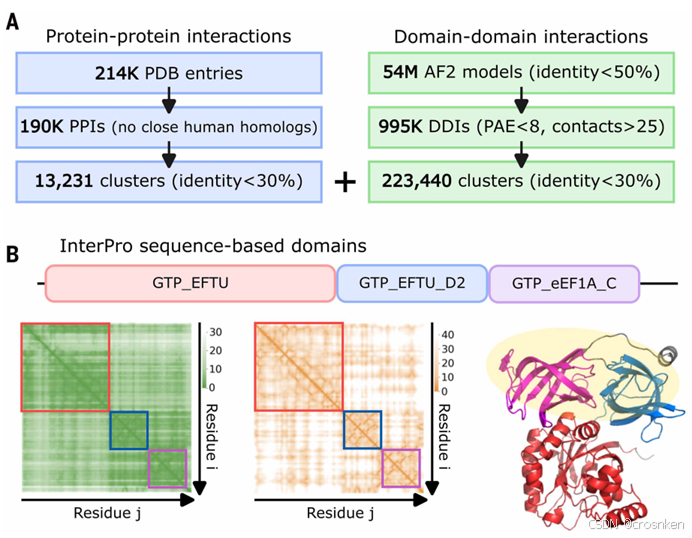
主要看A图右边,B图是interpro识别的domain与PAE识别的domain的对比
1、从AFDB筛选多域蛋白(至少包含两个Domain)
先用序列相似度50%进行蛋白质聚类,获得了54M个蛋白质类
2、DPAM识别结构域区间+已有的InterPro标注
利用AlphaFold预测结果中的PAE矩阵(衡量相对位置的不确定性),将PAE<8的区间聚类,然后将PAE差异过大的区间分到不同的domain中
3、提取蛋白内DDI
若同一蛋白质内的两个domain有至少25个残基接触( 𝐶𝛽距离小于6Å),则认为有DDI
4、对上述DDI再一次聚类,剔除掉与人类蛋白质组相似度高于30%的域,避免数据分布不均匀、数据泄露与过拟合
5、负采样
对每一个正样本在同物种中选择两个domain,验证在各个蛋白质中均不接触,且不被PPI并集中的蛋白质对分别包含。
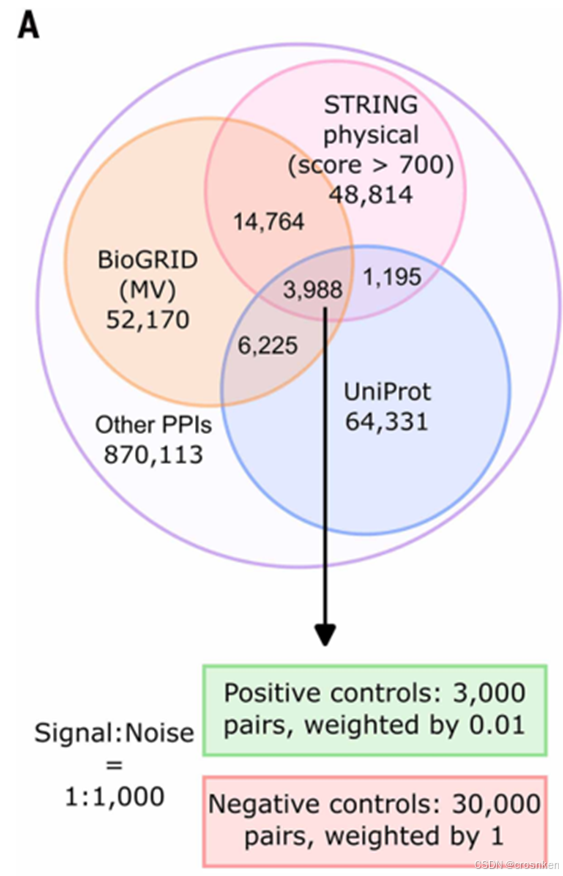
数据集构建
1、PPI数据集构建
从PDB数据库中,筛选异源二聚体结构,并判断两条肽链至少5个残基接触(𝐶_𝛽距离小于6Å),认定该对蛋白质具有PPI,将蛋白质以30%相似度聚类后,构建蛋白质类的相互作用网络。
剔除与人类蛋白质相似度高于30%的蛋白质类(避免数据泄露)
对每个类间相互作用,选择其中分辨率最高的二聚体结构PDB文件
负样本采样:随机采样
2、数据集总体结构
训练集(用AFDB构建的DDI、PDB提取的PPI)
蛋白质单体结构:正DDI:负DDI:正PPI:负PPI=2:1:1:1:1
测试集(人类物种,三个PPI数据库的交集)
正PPI:负PPI=1:10
测试集图:
RF2-PPI
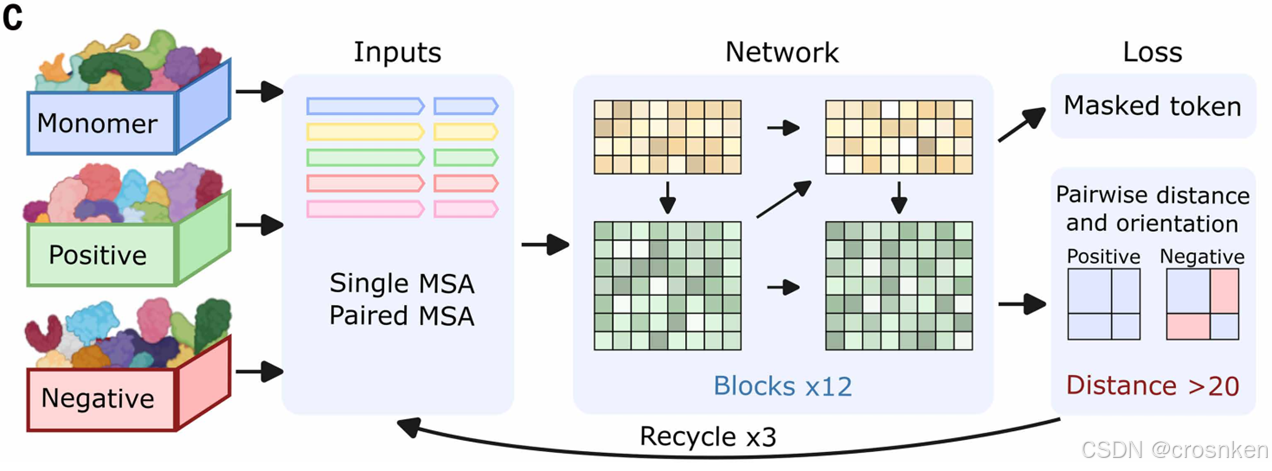
模型结构(具体可见参考材料M4.1)
嵌入层+简化RF2迭代+输出层
输入:
pMSA矩阵[n_species, seq_length(A+gap+B || A), onehot+label+PSSM] ,原序列[seq_length]
模型流程:
1、对pMSA和原序列分别做MLP和embedding,加起来,记为𝑚𝑠𝑎
2、构建残基间的pair features,𝑧_𝑖𝑗=𝐿𝑖𝑛𝑒𝑎𝑟(ℎ_𝑖⨂ℎ_𝑗),外积,线性层降维到𝑑_𝑧,记为𝑝𝑎𝑖𝑟
3、将上一次回收的𝑚𝑠𝑎和𝑝𝑎𝑖𝑟与当前的融合
4、12层RF2块迭代(行列注意力)+pair自身进行三角更新:𝑧_𝑖𝑗←𝑧_𝑖𝑗+∑_(𝑘=1)^(𝑠𝑒𝑞_𝑙𝑒𝑛)▒〖𝑧_𝑖𝑘⨀𝑧_𝑘𝑗 〗
5、输出网络
RF2-PPI迭代块
这里主要讲讲简化的RF2块:
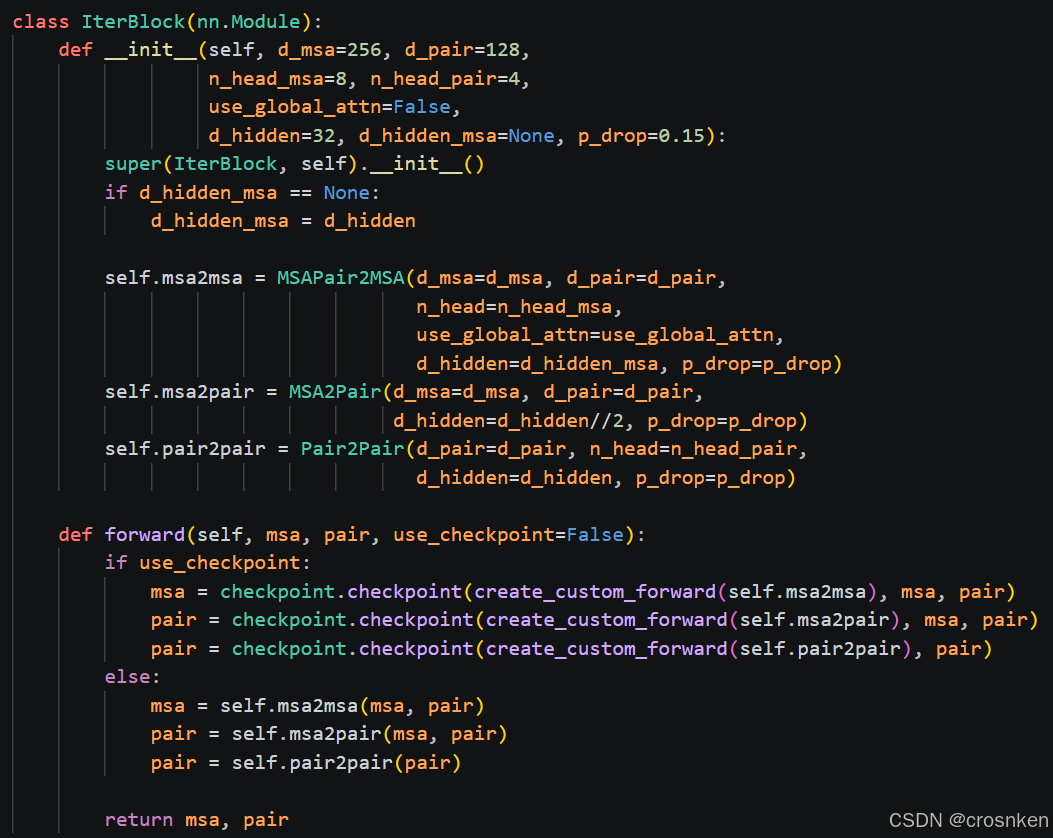
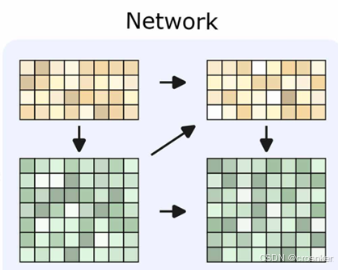
在RF2trackModule类中的IterativeSimulator中,每一个迭代块都包含三步:
计算右上的msa嵌入矩阵(MSAPair2MSA)
计算右下的pair特征矩阵(MSA2Pair)
对pair进行三角更新(Pair2Pair)
1、MSAPair2MSA,利用行列注意力交叉计算,在计算行注意力时,将pair的信息作为注意力矩阵的偏置注入进去:
# Update MSA with biased self-attention. bias from Pair
class MSAPair2MSA(nn.Module):
def __init__(self, d_msa=256, d_pair=128, n_head=8,
d_hidden=32, p_drop=0.15, use_global_attn=False):
super(MSAPair2MSA, self).__init__()
self.norm_pair = nn.LayerNorm(d_pair)
self.drop_row = CustomDropout(broadcast_dim=1, p_drop=p_drop)
self.row_attn = MSARowAttentionWithBias(d_msa=d_msa, d_pair=d_pair,
n_head=n_head, d_hidden=d_hidden)
if use_global_attn:
self.col_attn = MSAColGlobalAttention(d_msa=d_msa, n_head=n_head, d_hidden=d_hidden)
else:
self.col_attn = MSAColAttention(d_msa=d_msa, n_head=n_head, d_hidden=d_hidden)
self.ff = FeedForwardLayer(d_msa, 4, p_drop=p_drop)
def forward(self, msa, pair):
'''
Inputs:
- msa: MSA feature (B, N, L, d_msa)
- pair: Pair feature (B, L, L, d_pair)
Output:
- msa: Updated MSA feature (B, N, L, d_msa)
'''
B, N, L, _ = msa.shape
pair = self.norm_pair(pair)
# Apply row/column attention to msa & transform
msa = msa + self.drop_row(self.row_attn(msa, pair))
msa = msa + self.col_attn(msa)
msa = msa + self.ff(msa)
return msa2、MSA2Pair,没什么特别的,就是上面讲的外积+MLP
class MSA2Pair(nn.Module):
def __init__(self, d_msa=256, d_pair=128, d_hidden=32, p_drop=0.15):
super(MSA2Pair, self).__init__()
self.norm = nn.LayerNorm(d_msa)
self.proj_left = nn.Linear(d_msa, d_hidden)
self.proj_right = nn.Linear(d_msa, d_hidden)
self.proj_out = nn.Linear(d_hidden*d_hidden, d_pair)
self.d_hidden = d_hidden
self.reset_parameter()
def reset_parameter(self):
# normal initialization
self.proj_left = init_lecun_normal(self.proj_left)
self.proj_right = init_lecun_normal(self.proj_right)
nn.init.zeros_(self.proj_left.bias)
nn.init.zeros_(self.proj_right.bias)
# zero initialize output
nn.init.zeros_(self.proj_out.weight)
nn.init.zeros_(self.proj_out.bias)
def forward(self, msa, pair):
B, N, L = msa.shape[:3]
msa = self.norm(msa)
left = self.proj_left(msa)
right = self.proj_right(msa)
right = right / float(N)
out = einsum('bsli,bsmj->blmij', left, right).reshape(B, L, L, -1)
out = self.proj_out(out)
pair = pair + out
return pair3、Pair2Pair,这里的三角更新实际上在蛋白质结构预测模型中非常常用,是保持特征向量的几何约束的手段:
class TriangleMultiplication(nn.Module):
def __init__(self, d_pair, d_hidden=128, outgoing=True):
super(TriangleMultiplication, self).__init__()
self.norm = nn.LayerNorm(d_pair)
self.left_proj = nn.Linear(d_pair, d_hidden)
self.right_proj = nn.Linear(d_pair, d_hidden)
self.left_gate = nn.Linear(d_pair, d_hidden)
self.right_gate = nn.Linear(d_pair, d_hidden)
#
self.gate = nn.Linear(d_pair, d_pair)
self.norm_out = nn.LayerNorm(d_hidden)
self.out_proj = nn.Linear(d_hidden, d_pair)
self.d_hidden = d_hidden
self.outgoing = outgoing
self.reset_parameter()
def reset_parameter(self):
# normal distribution for regular linear weights
self.left_proj = init_lecun_normal(self.left_proj)
self.right_proj = init_lecun_normal(self.right_proj)
# Set Bias of Linear layers to zeros
nn.init.zeros_(self.left_proj.bias)
nn.init.zeros_(self.right_proj.bias)
# gating: zero weights, one biases (mostly open gate at the begining)
nn.init.zeros_(self.left_gate.weight)
nn.init.ones_(self.left_gate.bias)
nn.init.zeros_(self.right_gate.weight)
nn.init.ones_(self.right_gate.bias)
nn.init.zeros_(self.gate.weight)
nn.init.ones_(self.gate.bias)
# to_out: right before residual connection: zero initialize -- to make it sure residual operation is same to the Identity at the begining
nn.init.zeros_(self.out_proj.weight)
nn.init.zeros_(self.out_proj.bias)
def forward(self, pair):
pair = self.norm(pair)
B, L = pair.shape[:2]
left = self.left_proj(pair) # (B, L, L, d_h)
left_gate = torch.sigmoid(self.left_gate(pair))
left = left_gate * left
right = self.right_proj(pair) # (B, L, L, d_h)
right_gate = torch.sigmoid(self.right_gate(pair))
right = right_gate * right
if self.outgoing:
out = einsum('bikd,bjkd->bijd', left, right/float(L))
else:
out = einsum('bkid,bkjd->bijd', left, right/float(L))
out = self.norm_out(out)
out = self.out_proj(out)
gate = torch.sigmoid(self.gate(pair)) # (B, L, L, d_pair)
out = gate * out
return out
class Pair2Pair(nn.Module):
def __init__(self, d_pair=128, n_head=4, d_hidden=32, p_drop=0.25):
super(Pair2Pair, self).__init__()
self.drop_row = CustomDropout(broadcast_dim=1, p_drop=p_drop)
self.drop_col = CustomDropout(broadcast_dim=2, p_drop=p_drop)
self.tri_mul_out = TriangleMultiplication(d_pair, d_hidden=d_hidden)
self.tri_mul_in = TriangleMultiplication(d_pair, d_hidden, outgoing=False)
self.ff = FeedForwardLayer(d_pair, 2)
def forward(self, pair):
#_nc = lambda x:torch.sum(torch.isnan(x))
pair = pair + self.drop_row(self.tri_mul_out(pair))
pair = pair + self.drop_col(self.tri_mul_in(pair))
pair = pair + self.ff(pair)
return pair输出网络
输出网络:代码在AuxiliaryPredictor.py中
1、距离-角度网络
用𝑝𝑎𝑖𝑟计算残基间距离-角度分类,并确保距离和𝐶𝛼-𝐶𝛽-𝐶𝛽-𝐶𝛼 二面角对称性。
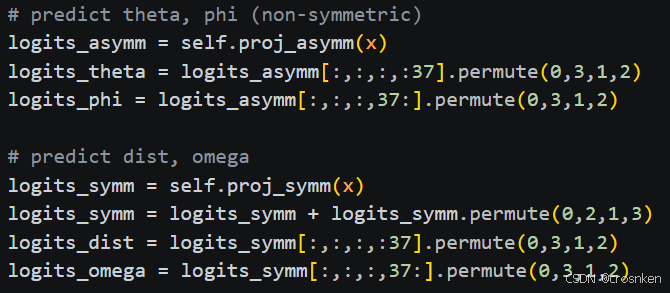
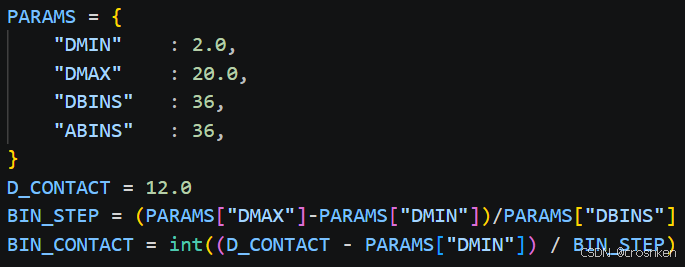
距离2Å~20Å,角度−π~π,分36箱,𝐶_𝛽距离20Å以外的残基单独分一个箱
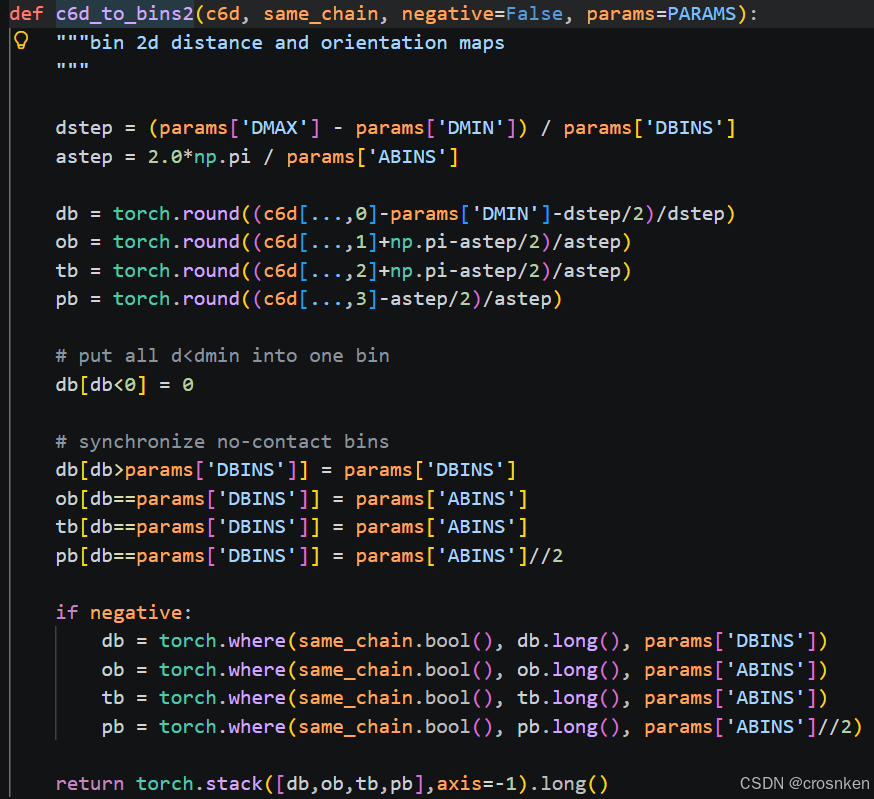
分箱处理在c6d_to_bins2函数中
采用交叉熵损失并求和
2、序列重构网络
用𝑚𝑠𝑎过一个线性层,重构氨基酸序列
采用交叉熵损失,只计算mask部分的平均损失
3、分辨率识别网络
如果残基中𝑁、𝐶𝛼、𝐶三个原子都没被mask,则将其视为高分辨率的残基
这里可以看mask_BB的构建与第五个损失函数的计算:
def _get_loss_and_misc(self, output_i, true_msa, mask_msa, true_crds, mask_crds, \
same_chain, eval_PPI=False, negative=False, return_bind=False):
logit_c6d_s, logit_aa_s, logit_exp = output_i
# processing labels for distogram orientograms
mask_BB = ~(mask_crds[:,:,:3].sum(dim=-1) < 3.0) # ignore residues having missing BB atoms for loss calculation
mask_2d = mask_BB[:,None,:] * mask_BB[:,:,None] # ignore pairs having missing residues
label_c6d = xyz_to_c6d(true_crds) # (B,L,L,4) 4->[dist,omega,theta,phi]
label_c6d = c6d_to_bins2(label_c6d, same_chain, negative=negative)# (B,L,L,37)x4
# calculate p_bind from predicted distogram
p_bind = self.active_fn(logit_c6d_s[0])[:,:BIN_CONTACT].sum(dim=1)
p_bind = p_bind*(1.0-same_chain.float()) # (B, L, L)
p_bind = nn.MaxPool2d(p_bind.shape[1:])(p_bind).view(-1)
p_bind = torch.clamp(p_bind, min=0.0, max=1.0)
#logits_bind = torch.log(p_bind) - torch.log(1-p_bind)
#loss, loss_s = self.calc_loss(logit_c6d_s, label_c6d, logits_bind, mask_2d, same_chain,
loss, loss_s = self.calc_loss(logit_c6d_s, label_c6d, p_bind, mask_2d, same_chain, logit_aa_s, true_msa, \
mask_msa, logit_exp, mask_BB, eval_PPI=eval_PPI, negative=negative, **self.loss_param)
if return_bind:
return loss, loss_s, p_bind
else:
return loss, loss_s
def calc_loss(self, logit_s, label_s, p_bind, mask_2d, same_chain, logit_aa_s, label_aa_s, \
mask_aa_s, logit_exp, mask_BB, eval_PPI=False, negative=False, w_dist=1.0, \
w_aa=1.0, w_exp=1.0, w_bind=0.0, eps=1e-6):
'''
Inputs:
- logit_s: 2D logits for distogram & orientogram [(B, C, L, L)*4]
- label_s: Labels for distogram & orientogram [(B, L, L)*4]
- mask_2d: 1 means Valid residue pairs (not including missing residues), (B, L, L)
- same_chain: whether two residues in the same chain (1 - same chain / 0 - different chain), (B, L, L)
- logit_aa_s: Logits for masked language modeling (MLM) task (B, C, N, L)
- label_aa_s: true labels for MLM task (B, N, L)
- mask_aa_s: masked positions for MLM task (1: masked, 0: not masked) (B, N, L)
- logit_exp: logits for predicting experimentally resolved region (B, L)
- mask_BB: whether they are experimentally resolved (1-resolved, 0-missing) (B, L)
- logits_bind: Logits for predicted probability to bind (B)
- eval_PPI: evaluate logits_bind or not
- negative: Whether given PPI is negative pairs or not
- w_*: weights for each loss terms
Outputs:
- Loss of the given predictions
'''
B, L = mask_2d.shape[:2]
loss_s = list()
tot_loss = 0.0
# col 0~3: c6d loss (distogram, orientogram prediction)
# for negatives, labels for inter-chain regions should assign as "FAR APART" (the last bin)
loss = calc_c6d_loss(logit_s, label_s, mask_2d)
tot_loss += w_dist * loss.sum()
loss_s.append(loss.detach())
# col 4: masked token prediction loss
loss = self.loss_fn(logit_aa_s, label_aa_s)
loss = loss * mask_aa_s
loss = loss.sum() / (mask_aa_s.sum() + eps)
tot_loss += w_aa * loss
loss_s.append(loss[None].detach())
# col 5: experimentally resolved prediction loss
loss = nn.BCEWithLogitsLoss()(logit_exp, mask_BB.float())
tot_loss += w_exp * loss
loss_s.append(loss[None].detach())
# col 6: binder loss
if eval_PPI:
if (negative):
target = torch.tensor([0.0], device=p_bind.device)
else:
target = torch.tensor([1.0], device=p_bind.device)
loss = torch.nn.BCELoss()(p_bind,target)
#loss = torch.nn.BCEWithLogitsLoss()(logits_bind,target)
tot_loss += w_bind * loss
else:
loss = torch.tensor(0.0, device=logit_s[0].device)
loss_s.append(loss[None].detach())
return tot_loss, torch.cat(loss_s, dim=0)将𝑚𝑠𝑎归一化后使用线性层分类,采用BCEWithLogitsLoss。
4、binder判定算法
用1中预测的距离矩阵,对分箱概率做Softmax,累和小于10Å的分箱概率,作为接触值
过滤掉同一蛋白质中的接触值后,找到最大的接触值,作为最终的PPI、DDI预测结果
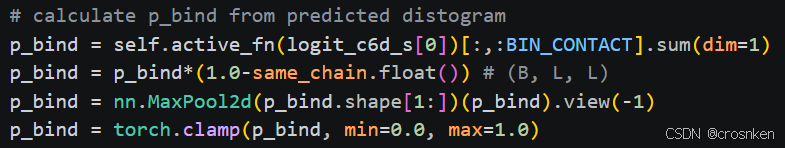
损失函数采用BCELoss
训练过程
训练集数据占比→蛋白质单体:正DDI:负DDI:正PPI:负PPI=2:1:1:1:1
PDB单体蛋白质结构(2020.4.30之前)→正PPI→负PPI→facebook的蛋白质结构(可能是ESMFold预测结构)→正DDI→负DDI
训练过程在DistilledDataset的构建中可以看到。
训练141轮,batch_size默认1,每轮总共采样25600条数据
采样数据量更改在DistributedWeightedSampler的参数中。
采用指数移动平均更新(EMA)(平滑训练时的模型权重)
RF2-PPI结果分析
测试集PPI正负样本比1:10
评价指标:带权PR曲线
𝑝𝑟𝑒𝑐𝑖𝑠𝑖𝑜𝑛=(𝑇𝑃∗0.01)/(𝑇𝑃∗0.01+𝐹𝑃), 𝑟𝑒𝑐𝑎𝑙𝑙=𝑇𝑃/(𝑇𝑃+𝐹𝑁)
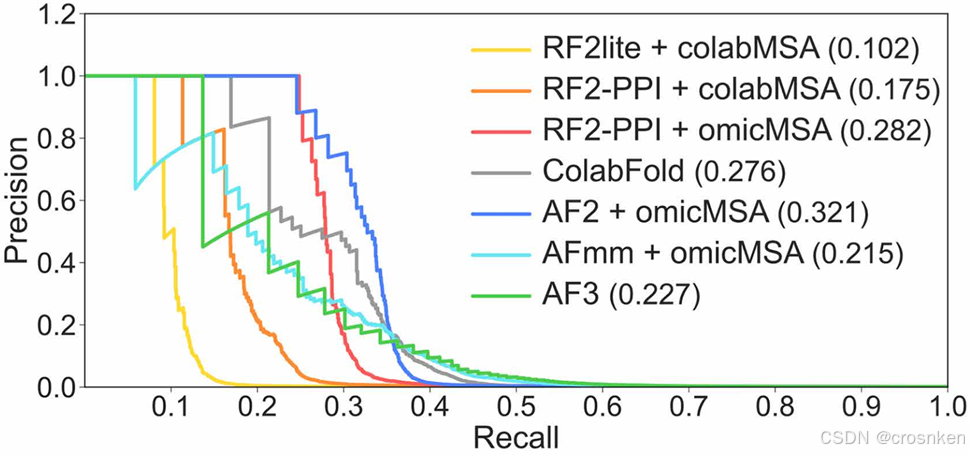
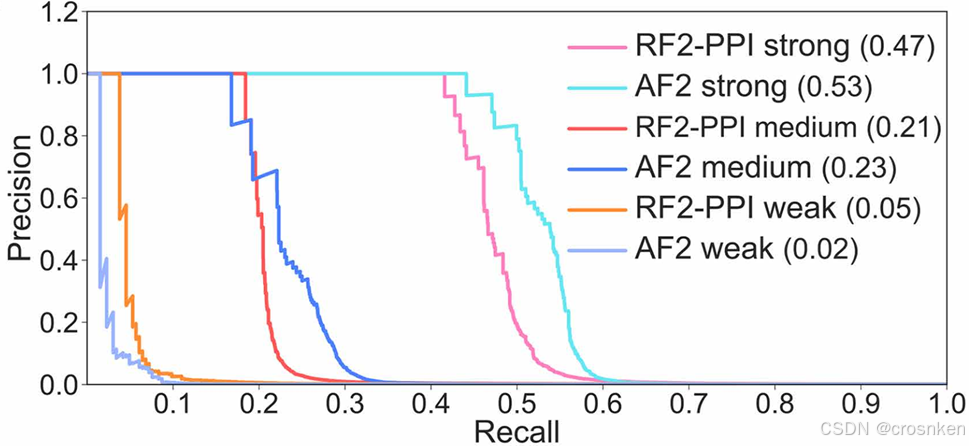
然后与AF2+omicMSA做对比,测试了三类PPI上的预测性能
强PPI:接触残基数大于50
中等PPI:接触残基数在11到50之间
弱PPI:接触残基数小于等于10
相比于AF2,能够发现更弱的PPI关联
AF2精筛
首先整理一下PPI筛选的流程
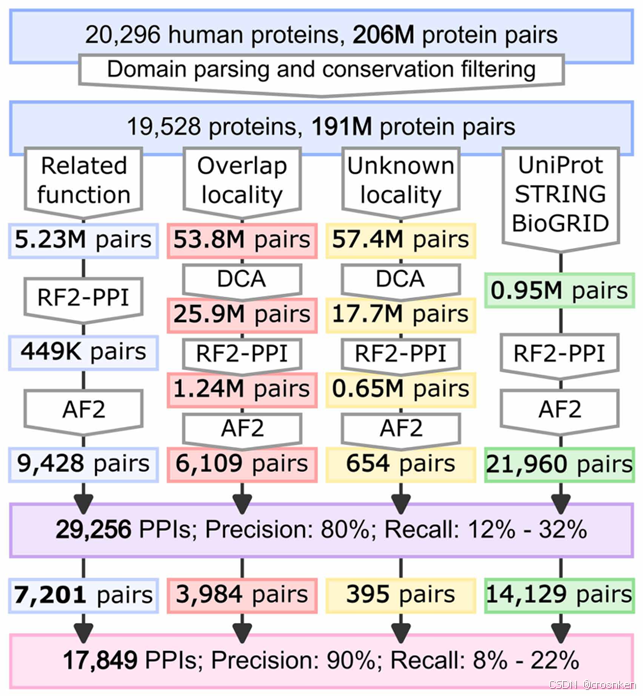
1、基础筛选
去掉过长或过短的蛋白质、去掉无序结构的蛋白质
去掉pMSA中共进化物种数少于10的蛋白质对
2、分类
(1)对数目较多的类进行DCA预筛选
DCA:两个位点的耦合系数矩阵的F-范数,取top-k求平均
耦合系数:pMSA中,两个位点对的氨基酸频率协方差矩阵的逆
𝐶_𝑖𝑗 (𝑎,𝑏)=𝑓_𝑖𝑗 (𝑎,𝑏)−𝑓_𝑖 (𝑎)𝑓_𝑗 (𝑏),对𝐶求逆元
(2)利用BP和CC标签进行筛选
分三类:具有相关的BP标签,具有重叠的CC标签,CC标签未知
3、使用RF2-PPI预测,阈值为0.5
4、使用AF2进一步筛选
这一段就是经典的AF2 PPI分析的套路了
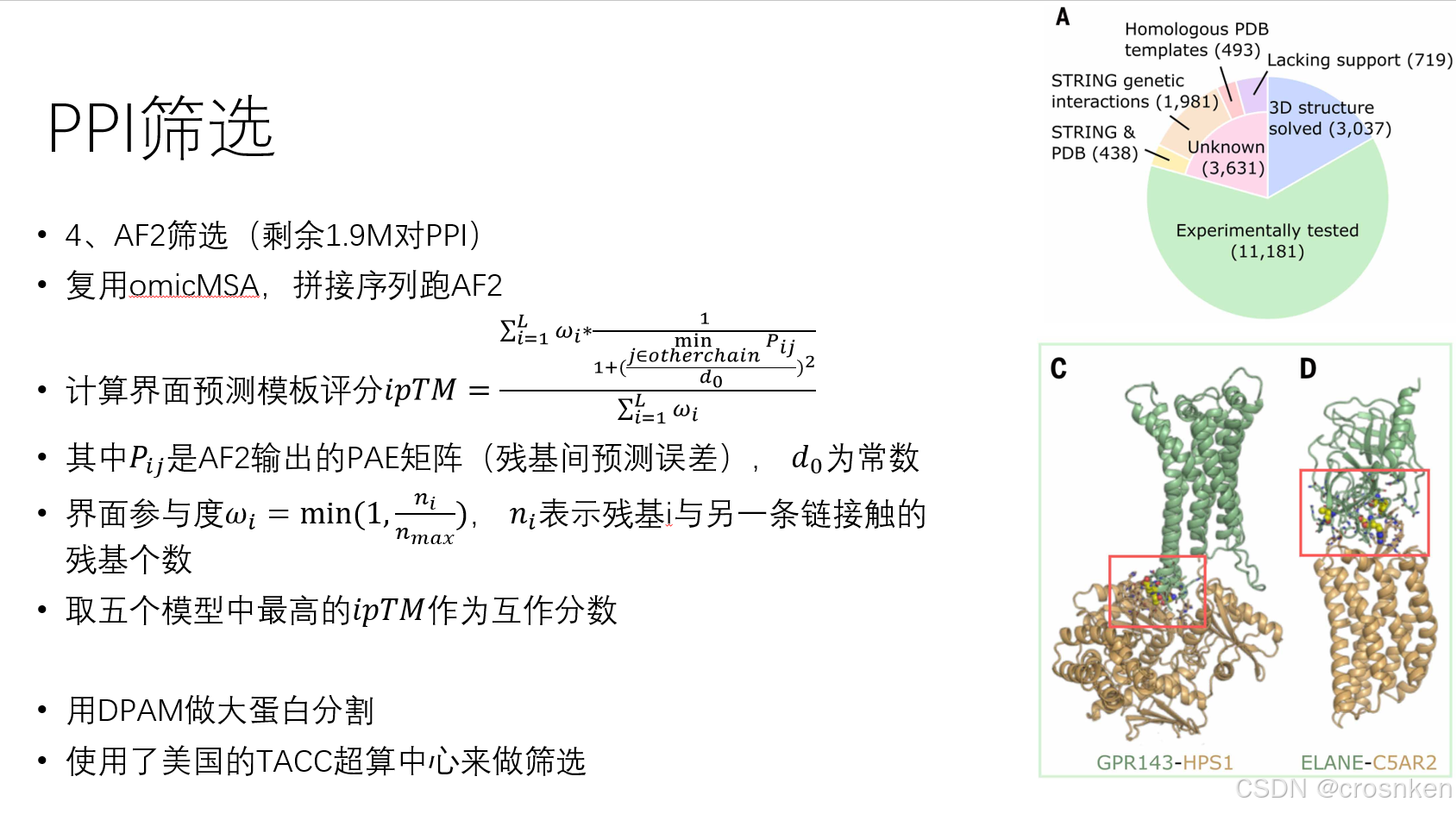
最后附上论文中精美的的结果图:
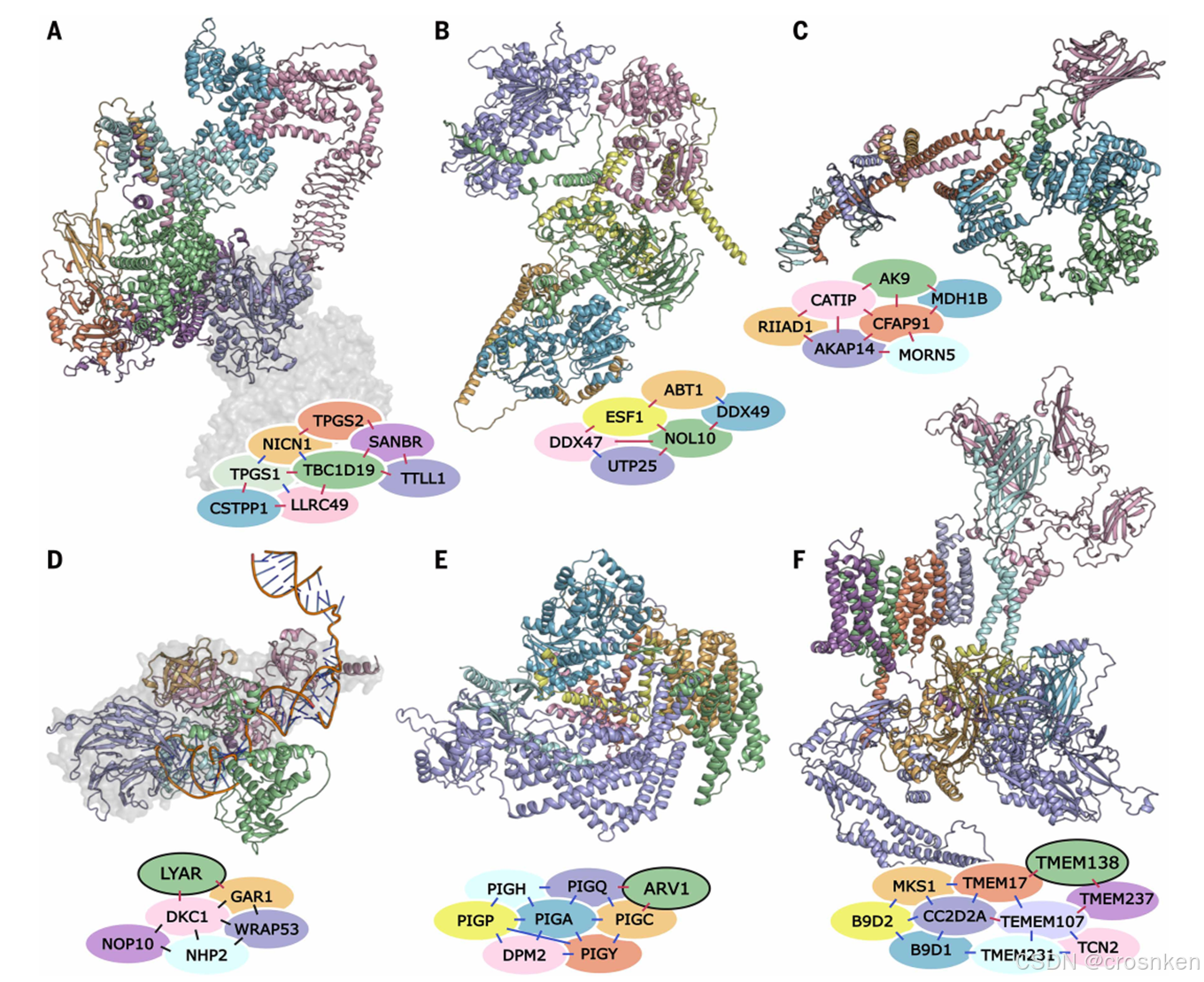
结语
个人评价:传统生信与深度学习结合的大成之作
omicMSA构建以及最后的AF2精筛包含了极度庞大工作量,DDI挖掘的创新以及RF2-PPI中的网络结构简化,在极端信噪比1:1000情况下,PPI预测准确度依旧惊人。
代码结构十分优雅,赏心悦目。
后续论文中还进行了一系列的生物学分析,包括GPCR预测、人类免疫系统蛋白质研究、线粒体靶向蛋白的PPI研究、高阶蛋白质复合物研究,并非只是简单的指标SOTA就发论文,而是真真实实地在用预测推动科研。

AtomGit 是由开放原子开源基金会联合 CSDN 等生态伙伴共同推出的新一代开源与人工智能协作平台。平台坚持“开放、中立、公益”的理念,把代码托管、模型共享、数据集托管、智能体开发体验和算力服务整合在一起,为开发者提供从开发、训练到部署的一站式体验。
更多推荐


 8
8 0
0- 0



 已为社区贡献1条内容
已为社区贡献1条内容

所有评论(0)