【通路信息先验+多组学+有开源代码】DeePathNet:基于Transformer的深度学习模型,将多组学数据与癌症通路整合
论文总结
1、提出了将基因组突变、拷贝数变异、基因表达、DNA甲基化、蛋白质强度和CRISPRCas9数据等多组学数据整合的模型,并且引入了通路信息,先将多组学特征embedding成pathway级别特征,然后再是Transformer和MLP层。并且在药物反应、癌症分型、乳腺癌亚型分类等任务上验证模型性能。整体来说模型结构比较简单,优势在于降低维度、避免维数灾难和可解释性强。
2、有开源代码:https:// github.com/CMRI-ProCan/DeePathNet
3、对于多组学数据整合,在基因层面把不同组学数据拼接在一起。每个通路的基因特征被输入到一个独立的全连接层中,生成该通路的表示向量(维度512),通过可学习的权重将多组学信息整合为通路级表示。这些通路表示再通过Transformer进行通路间交互建模,实现更高层次的融合。
4、消融研究中,比较了“通路替换为随机基因”和“去掉Transformer”的变体,证明了通路信息和Transfor架构对模型性能均有贡献,特别在数据量较少时,通路先验更加有用。
摘要
结合机器学习的多组学数据分析有望显著改善癌症诊断和预后。传统机器学习方法通常仅限于组学测量,省略了现有领域知识,如连接不同组学数据类型中分子实体的生物网络。在此,我们开发了一个基于Transformer的可解释深度学习模型DeePathNet,将癌症特异性通路信息整合进多组学数据分析中。利用包括ProCan-DepMapSanger、癌症细胞系百科全书和癌症基因组图谱在内的多种大数据集,我们证明并验证了DeePathNet在预测药物反应及癌症类型及亚型分类方面优于传统方法。结合生物医学知识和最先进的深度学习方法,DeePathNet使得通路层面的生物标志物发现成为可能,最大化数据驱动癌症研究方法的力量。DeePathNet 可在 GitHub 上访问,网址为 https:// github.com/CMRI-ProCan/DeePathNet。
重要性:DeePathNet 利用基于变换器的深度学习整合癌症特异性生物通路,增强癌症分析能力。它在预测药物反应、癌症类型及亚型方面优于现有模型。通过实现通路级生物标志物的发现,DeePathNet 代表了癌症研究的重大进展,有望带来更有效的治疗方法。
引言
对不同数据类型的多组学分析使研究人员能够深入了解肿瘤生物学,并识别新的且稳健的治疗靶点(1)。机器学习多组学分析的一个主要目标是预测最适合精准医疗背景下的癌症治疗策略。多种多组学研究改善了肿瘤内异质性的检测、新治疗靶点的识别以及更稳健的诊断和预测标志(2–5)。许多发现仅靠分析单一的组学数据类型是不可能实现的。然而,由于高通量仪器产生的大量数据以及现有多组数据整合方法的局限性,进行多组分析面临计算挑战(6, 7)。
为此,开发了大量机器学习方法用于集成大规模多组数据(2–4, 7–11)。例如,moCluster(8)集成基于联合潜变量模型的多组数据,性能优于之前的方法如iCluster(9)和iCluster Bayes(10)。同样,mixOmics(11)提供了多种多组数据集成选项,旨在寻找不同组学数据类型的共同信息。基于变分自编码器的方法也被应用于多组积分(bioRxiv 2024.06.26.600742;参考文献12,13)。例如,scVAEIT(12)对单细胞多模态数据集成和补值进行了稳健的概率建模,MOVE(13)利用生成式深度学习模型发现了2型糖尿病中的药物-组学关联。然而,这些模型主要关注学习数据驱动的表征,未考虑将不同组学数据类型联系起来的现有生物医学知识,如调控网络。 深度学习已被广泛应用于癌症研究中的类似任务,展示了更优的预测性能和捕捉高维数据复杂模式的能力。例如,深度学习模型已被用于整合多组数据以预测肝癌患者的生存率(14),从癌症转录组中提取生物学相关的潜伏空间(15),并将组织学图像与基因组数据结合以预测癌症结局(16)。尽管这些方法突出了深度学习在癌症研究中的潜力,但它们主要依赖数据驱动的方法,未纳入现有的生物医学知识,如通路或调控网络。监管网络存在于细胞中,通过功能相互作用的蛋白质或RNA大分子集合来调控各种基因产物的表达水平(17)。然而,结合现有生物医学知识和计算推断的模型,有潜力更好地捕捉驱动生物标志物关联的相互作用,并提升这些算法的预测能力和建模能力(7)。 在组学研究中,大多数现有将生物医学知识融入机器学习的尝试仅限于单一组学数据类型(18–21页)。ATHENA(22)和PARADIGM(23)支持多组数据,但它们基于线性模型,缺乏足够复杂以建模路径间关系。利用深度学习,多项研究尝试将现有生物医学知识整合到多组学模型中(3, 24)。DCell(25)和DrugCell(26)将神经网络架构与已知的基因本体信息结合,但它们仅支持使用基因缺失或突变作为输入。EMOGI(27)基于图神经网络(GNN;参考文献28、29)设计,将蛋白质-蛋白质相互作用网络与多组数据整合,用于预测癌症基因。然而,其网络架构难以轻易推广到其他任务。此外,基于GNN的模型被开发用于利用多组数据分类乳腺癌亚型,但该模型无法用于发现潜在的新生物学机制或药物靶点(30)。同样,通路模型在癌症研究中被广泛应用,以加深对肿瘤生物学的理解并改进预测模型。例如,像PARADIGM(23)和NetGSA(31)这样的方法整合了通路信息,从基因组数据推断通路活动并进行差异性通路分析。尽管这些方法利用了通路知识,但癌症特异性通路模型尚未被纳入多组数据集成深度学习模型的设计中。将癌症通路(32)整合进深度学习的多组学数据分析中,用于药物反应预测和癌症类型或亚型分类等一般任务,仍是一个开放的研究课题。 受生成预训练变换器(2018年 hayate-lab.com;https://hayate-lab.com/wp-content/uploads/ 2023年5月43372bfa750340059ad87ac8e538c53b.pdf)对ChatGPT(33)近期成就做出重大贡献的计算基础启发,我们开发了DeePathNet,这是一种基于Transformer(34)的可解释深度学习方法,整合了多组数据,如基因组突变、拷贝数变异、基因表达、DNA甲基化、蛋白质强度和CRISPRCas9数据,包括癌症通路知识。Transformer架构推动了人工智能领域的许多突破(35–37)。在分子生物学中,该变换器已被用于建模DNA序列、蛋白质三级结构和药物化学结构数据(bioRxiv 2023.01.11.523679;参考文献38、39)。此外,基于变压器的模型与GNN结合使用,用于分析癌症中的mRNA和miRNA数据,但这些模型无法被解释(40, 41)。因此,目前尚不清楚该变换器是否能与癌症通路信息结合,集成任意数量的组学数据层进行多组癌症数据分析。
我们在本文中的贡献可以总结如下:
1. 开发了一种名为DeePathNet的新颖深度学习模型,用于分析癌症分子数据。DeePathNet的创新之处在于其独特设计,使Transformer能够解决癌症研究中此前未解之谜。通过将变换器与癌症路径的领域特异性知识结合,DeePathNet不仅超越了传统机器学习方法的预测精度,还提供了可靠的模型解释。
2. DeePathNet的性能已通过大规模数据集和多种评估指标进行评估和验证,这些指标在类似研究中并不常见。由于拥有相对庞大的数据,被选定了包括药物反应预测以及癌症类型和亚型分类在内的典型任务。DeePathNet 可以通过调整输出神经元数量扩展到其他任务。
3. 利用DeePathNet的特征重要性报告了主要的组学特征和癌症通路,促进了潜在新生物标志物的发现。这些信息为了解癌症发展和进展的潜在机制提供了宝贵见解。
材料和方法
多组学和药物反应数据收集
为预测药物反应,检索了941个细胞系项目(CLP;参考文献42)和696个癌细胞系百科全书(CCLE)细胞系(43)的多组数据。CLP中共有19,099个基因突变、19,116个拷贝数变异(CNV)和15,320个基因表达特征,CCLE中包含18,103个基因突变、27,562个CNV和19,177个基因表达特征。 在蛋白质组学数据的药物反应预测分析中,加入了 CLP 的 ProCanDepMapSanger 数据集(44)(CLP + ProCanDepMapSanger 1⁄4 CLP+),同时也使用了 CCLE 的蛋白质组数据集(45)(CCLE + CCLE 蛋白质组数据 1⁄4 CCLE+)。ProCan-DepMapSanger 和 CCLE 蛋白质组学数据集分别包含 8,498 和 12,755 个蛋白质特征。合并后的数据集分别包含910个细胞系和292个CLP+细胞系和CCLE+。未对数据集进行额外处理(补充表S1)。在使用CCLE和CCLE+作为独立测试集时,我们排除了CLP数据集中同样存在的细胞系,只使用了CCLE和CCLE+数据集中独特的71株和33株细胞系。 在癌症类型和亚型分类方面,使用TCGA组装器2(46)检索了癌症基因组图谱(TCGA)队列的多组数据。共收集了6,356个样本,包含31,949个基因突变特征、23,529个CNV特征和20,435个基因表达特征。此外,还检索了来自临床蛋白质组肿瘤分析联盟(CPTAC)乳腺癌队列(47)的122个乳腺癌样本的多组数据,包含11,877个基因突变特征、23,692个CNV特征和23,121个基因表达特征。对于乳腺癌亚型分类,检索了微阵列50预测分析(PAM50)分类(腔内A、腔内B、HER2+、基础和正常样)数据(补充表S1)。 当某些数据模态(例如基因表达)无法提供时,我们通过在输入向量中插入零来表示缺失值。这种方法实际上排除了对应神经元在训练过程中对模型预测的影响,因为零输入不会影响神经元的激活。因此,这些神经元在权重更新时会被忽略,使模型能够处理缺失数据而无需补补或排除整个样本。
DeepPathNet概述
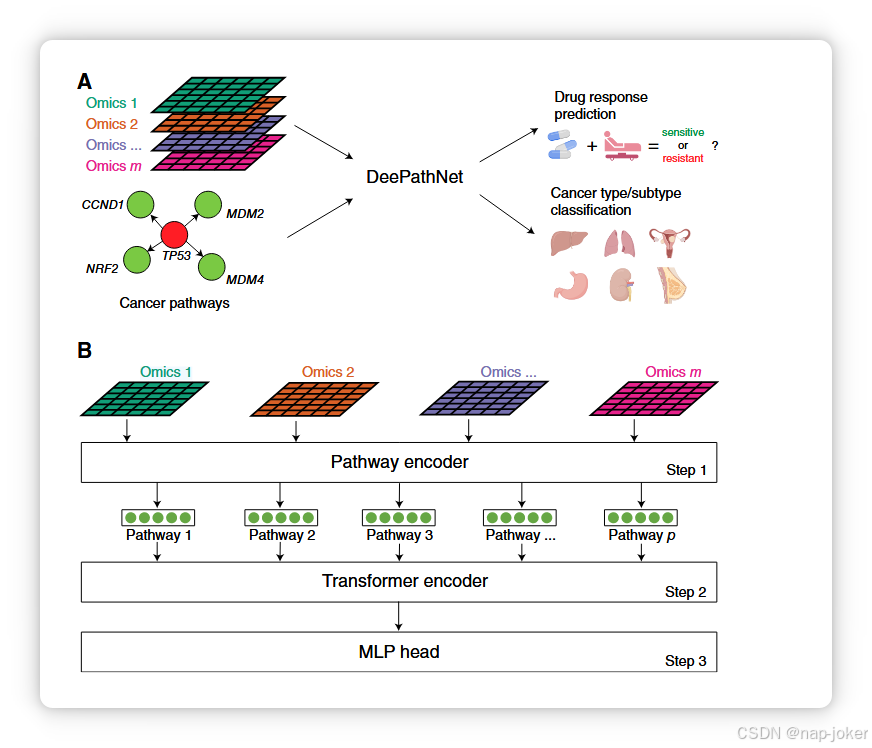
图1 DeePathNet概述。DeePathNet的网络架构基于LCPathway数据集构建,并以多组学数据为输入,用于建模通路相互作用、预测药物反应或分类癌症类型和亚型。B,DeePathNet架构支持任意数量的omic数据类型作为输入。步骤1:DeePathNet将多组信息编码到癌症通路中。步骤2:DeePathNet使用变压器编码器学习这些路径之间的相互作用。步骤3:将编码的路径向量传递到MLP进行预测。圆形表示神经网络中的神经元。箭头代表信息流的方向。(A,用 BioRender.com 创建。)
DeePathNet 旨在利用基于变换器的深度学习架构建模生物通路,输入包括多组数据和癌症通路信息(图1A)。DeePathNet在药物反应预测以及癌症类型和亚型分类方面进行了评估。 DeePathNet 包含三个主要步骤。它从一个通路编码器开始,将从组数据类型中总结特征到癌症通路(步骤1;图1B),然后使用变压器编码器模拟这些通路之间的相互作用(步骤2)。随后是多层感知器(MLP),可适应不同的预测任务(步骤3)。 在第一步中,神经网络架构基于LCPathways数据集(32)构建,该数据集包含241条文献精选通路,涵盖3164个癌症基因。LCPathways数据集之所以被选中,是因为它是最新且最全面的癌症研究路径数据库之一,专门为癌症研究精心策划。因此,它特别适合DeePathNet的应用。通路编码器随后使用全连接层,将基因(基因1 n)的多组数据(组学1 m)投射到代表癌症通路之一的512维通路载量(通路1 p;参见“材料与方法”;补充图S1A)。通过这种架构,路径编码器允许DeePathNet捕捉不同omic数据类型的交互。此外,将多组特征分组为通路会降低原始高维数据的维度。这缓解了维度诅咒的挑战,使得在计算上可行地将模型应用于大型数据集而不牺牲其性能(48)。 在第二步中,开发了增强版变压器模块,用于编码癌症通路之间的相互作用(参见“材料与方法”;补充图S1B)。首先,每次迭代仅训练一半路径,防止模型聚焦于无法很好地推广到测试数据集的特定路径。然后,使用原始变换器模块(34)中的两个模块,其中包含一个重复出现的层列表,每层包含层归一化、多头自注意和一个MLP序列。变换器还支持对癌症通路之间复杂关系的动态建模,从而避免了传统机器学习中为不同输入生成固定权重的情况。 在第三步,使用MLP将编码的通路向量映射到输出神经元,这使得变换器模块学到的知识能够适应通用的预测任务。DeePathNet的三个组成部分协同工作,无法单独应用于多组数据。
DeePathNet 详情
DeePathNet 有路径编码器(步骤1)、变换器编码器(步骤2)和MLP(步骤3)。每个基因的输入载体是所有可用数据模态特征的串接。例如,如果有突变、CNV和RNA数据,则基因i的输入载体 习 构造为 习 1⁄4 1/2 x突变 i;xiCNV;xiRNA 中,每个x模态i代表该模态的特征值。如果某个基因缺少一个模态,我们会在xmodality i中插入一个零。 在第一步中,DeePathNet将多组信息编码到癌症通路中,这些通路由LCpathways中的241条癌症通路定义(32)。设变变∈ f0;1g代表突变,gCNV∈为CNV,gRNA为基因表达∈,gprot∈为基因g的蛋白质强度。那么向量包含n个基因且具有四种Omic数据类型的通路的OMIC特征定义为:
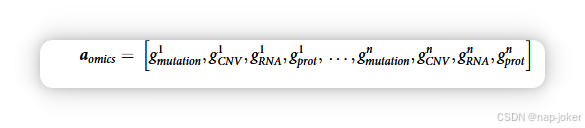
接着,向量aomics被编码到通过MLP编码的通路向量中。这里,符号转换为矩阵形式,以包含样本数。因此,对于N个样本,一条路径的四种omic数据类型的总特征可以表示为N 4n维的矩阵Aomics。DeePathNet随后使用全连通层将这些组学特征编码为编码路径矩阵Aencoded,计算公式为:
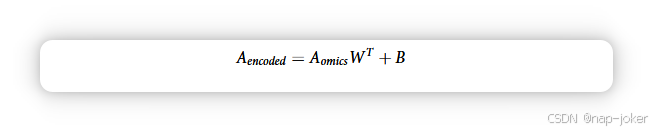
其中 W 和 B 分别代表全连通层中的可学习权重矩阵和偏置项。权重矩阵W的维数设为512* 4n,编码矩阵和B的维数均为N*512。共使用了241条癌症通路,编码了241个矩阵A1;A2编码;. . . ;编码的A241被组合为一个张量,Aen编码的维数为N 512 241。Aencoded 作为变压器编码器的输入(补充图 S1)。 在第二步,DeePathNet 使用变压器编码器来学习癌症调控通路之间的相互依赖性。与模拟输入与目标相互依赖的通用注意力机制不同,变换器模块利用自注意来模拟输入内部的相互依赖关系(即多组数据中的特征;参考文献49)。变压器编码器从241条癌症通路中概率为0.5的脱落层开始,确保训练过程中平均有一半通路被脱落,以防止潜在的过拟合。每个训练批次独立采样所选通路,允许使用不同的通路。变压器模块的配置与原始版本相同(34),下文称为变压器。由于变换器编码器包含循环层,我们使用带括号的上标表示不同层的编码,其中 Að0Þ 编码表示进入第一层之前的数据。变压器块第一层之后,Að0Þ 编码为 Að1Þ,编码方式如下:
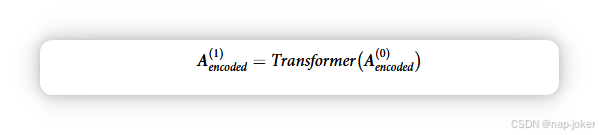
DeePathNet包含两层Transformer模块,因此:
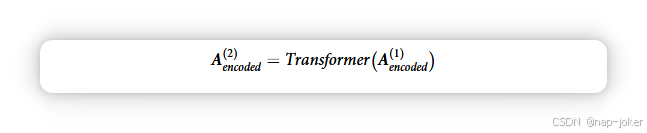
最后,在第三步,DeePathNet 使用 MLP 映射 A(2)_encoded,并将其编码为最终预测。MLP的输出维度取决于预测任务。对于药物反应预测,输出维度数等于药物数量,而对于癌症类型和亚型分类,输出维度数等于癌症类型和亚型的数量。
模型训练
随机森林模型以n_estimators =100和基尼不纯度为分裂准则进行训练。弹性网模型以α =1训练,l1_ratio = 0.5,迭代1000次。对于主成分分析(PCA)模型,采用了前200个主要成分。k-最近邻(k-NN)模型训练时使用k = 5。其他细节,包括其他比较方法的默认设置,可以在 Scikit-Learn(v1.0.2,RRID: SCR_002577)的官方 API(v1.0.2)中找到。对于混合组学,block.spls 模式用于多组数据集成,ncomp 对应每个组数据类型为 50。对于 moCluster,使用 mbpca 函数,ncomp= 200,k=“all”,method = “globalScore”,选择“lambda1”。由于PCA、moCluster、MOVE和scVAEIT都是无监督方法,它们能降低数据的维度,但不单独执行分类或回归任务,我们使用随机森林作为下游预测模型。具体来说,在将这四种方法应用于多组数据以获得低维表示后,我们对这些特征训练了一个随机森林分类器以执行监督预测任务。这种方法使我们能够结合稳健分类算法,评估PCA、moCluster、MOVE和scVAEIT提取特征的预测能力。用于 DeePathNet 的超参数可以在 GitHub 仓库中找到。为了训练DeePathNet进行回归,计算了预测值与实际IC50之间的均方误差损失。为了分类,我们计算了交叉熵损失以训练DeePathNet。 为应对深度学习样本量有限的挑战,未对每种方法进行超参数调优。进行全面的网格搜索需要保留部分数据集用于最终性能评估,从而减少训练集规模,并可能影响模型性能。鉴于观察到的微小变化和大量超参数调优所需的时间,所有方法均使用默认超参数进行训练。这一决策确保了不同模型之间的一致性比较,同时避免了个性化超参数优化带来的额外变异。 DeePathNet的计算时间见补充表S2,仅与随机森林网和弹性网进行了比较。
消融研究详情
我们对药物反应预测、癌症类型分类和乳腺癌亚型分类这三项任务进行了消融研究。研究比较了DeePathNet与两个对照组的表现:(i)仅变换器模型,该模型中癌症通路被随机选择的基因替代;(ii)无癌症通路或变换器结构的普通神经网络。
训练和测试数据
所有任务均使用相同的数据集以确保比较一致性。在药物反应预测任务中,使用CLP数据集进行训练,CCLE数据集作为独立测试集。在癌症类型分类方面,使用了来自6,356个TCGA样本的多组数据,并采用了5重交叉验证,将数据集分为训练集和测试集。最后,在乳腺癌亚型分类任务中,使用TCGA数据集中974个乳腺癌样本的多组数据进行训练,而独立的CPTAC队列(共122个样本)作为测试集。
仅有Transformer模型的创建
仅变换器模型是通过用随机选择的基因替换DeePathNet中生物筛选的癌症通路生成的。仅Transformer模型中的通路设计与原始通路大小相匹配,但不考虑生物学相关性。该随机化共进行了10次,以减少任何单一随机配置的影响。同样的DeePathNet架构也应用于这些随机通路,使得仅变换器模型与生物学意义上的通路之间可以直接比较。
评估指标
所有任务均采用相同的指标进行绩效评估。在药物反应预测方面,评估指标包括R²、平均绝对误差(MAE)和皮尔逊相关系数。对于癌症类型分类及乳腺癌亚型分类,指标包括准确率、宏观平均F1分数、ROC曲线下的面积(AUROC)和精确回忆曲线下的面积(AUPRC)。这些指标分别计算了独立测试集(CCLE用于药物反应,CPTAC用于乳腺癌亚型)以及TCGA数据集上的交叉验证(用于癌症类型分类)。
数据的可利用性
本研究所用的所有数据均为公开数据集。数据可从引用的原始出版物中下载。具体来说,https://cellmodelpassports。sanger.ac.uk/downloads 用于CLP和CLP+,https://depmap.org/portal/ 用于CCLE数据,https://github.com/BioinformaticsFMRP/TCGAbiolinks 用于TCGA数据,https://proteomic.datacommons.cancer.gov/pdc/ 用于CPTAC数据。DeePathNet的源代码和文档可在 https://github.com/CMRIProCan/DeePathNet 获取。源代码、处理后的数据集和中间文件也可在 https://doi.org/10.6084/m9.figshare.24137619 获取。
结果
DeePathNet预测药物反应
我们首先通过与随机森林(50)、弹性网(51)、PCA、mixOmics(4)、moCluster(8)、MOVE(13)和scVAEIT(12)进行基准测试,评估DeePathNet在回归任务中的预测表现,以预测抗癌药物对癌细胞系的反应。这八种方法采用了CLP(42)和CCLE(43)的数据进行评估,这两大是目前公开的多组癌细胞系数据集(参见“材料与方法”;补充表S1)。DCell(25)、DrugCell(26)和EMOGI(27)未纳入基准测试,因为它们不支持多组数据和一般预测任务。输入数据包括基因突变、CNV和基因表达数据。关于药物反应数据,我们检索了癌症药物敏感性基因组学(GDSC;参考文献42)数据库中的IC50。对每种方法,评估了六个实验设置,包含两个数据集和三个评估指标,即确定系数(R²)、MAE和Pearson相关系数(Pearson相关系数r),即预测值与实际IC50值之间的关系(参见补充材料和方法)。 在DeePathNet中,构建了241个通路编码器(补充图S1A),将组学数据汇总为LCPathways定义的通路向量(32)。这些载体随后被输入变压器模块,以模拟癌症通路之间的相互作用(补充图S1B)。所有八种方法都使用默认超参数(参见“材料与方法”)。组学数据通过早期积分(7)对随机森林和弹性网进行了合并。中间积分(7)用于PCA、移动集群和混合组学。PCA和moCluster与随机森林进行预测(参见“材料与方法”;参考文献7)。
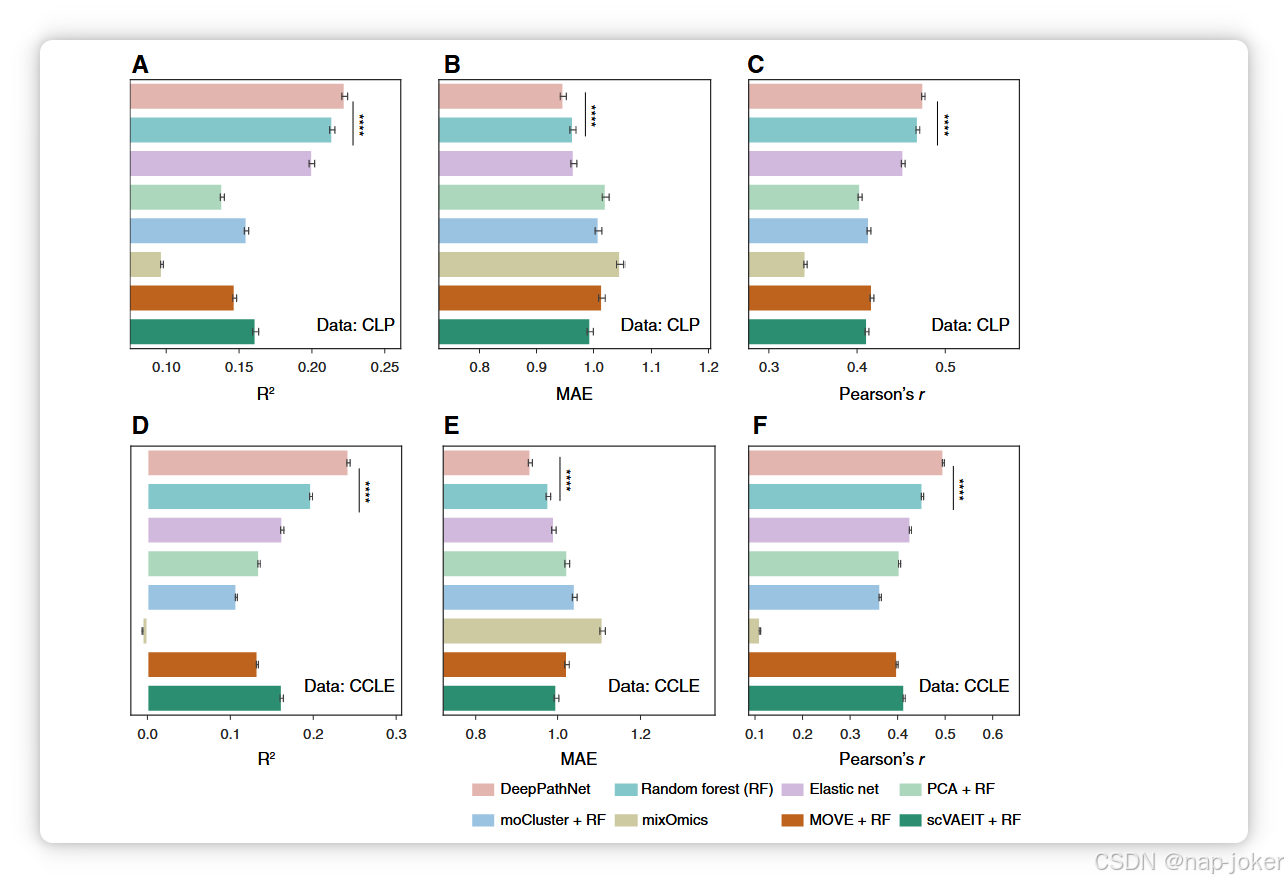
图2 通过交叉验证对药物反应预测的性能评估。A–F,条形图展示了六个实验设置在CLP和CCLE数据集上的预测性能,采用三个评估指标:R2(越高表示更准确),MAE(越低越准确),Pearson's r(越高越准确)。误差条由交叉验证得出,代表均值的95%置信区间。, P<0.0001,通过双尾配对学生t检验,仅显示第一和第二表现方法之间的显著性。这八种方法在底部用颜色标注。
为定量且可靠地比较这八种方法,随机重复五次交叉验证,分别对R2、MAE和皮尔逊r指标各得出25个误差指标。报告了评估指标的平均值和95%置信区间(CI),作为泛化误差的估计值。我们观察到,DeePathNet在药物反应预测方面表现显著且持续优于其他七种不包含癌症通路信息的方法(见图2AF;P值<0.0001;双尾配对学生t检验;补充表S3)。通过根据每种设置的平均值指标对方法进行排名,我们发现随机森林是表现第二好的方法。为了评估DeePathNet难以预测的药物反应是否在其他方法中同样具有挑战性,我们计算了DeePathNet预测与其他七种方法预测之间的相关性。我们观察到DeePathNet的预测表现与我们评估的其他七种方法高度一致,在不同方法间显示一致一致(Pearson's r > 0.9)。DeePathNet整体表现略优,反映在其多项指标上的持续更高准确率(补充图S2A和S2B)。这种性能一致性表明,与这些既有方法相比,DeePathNet没有产生任何意外或异常的预测。我们发现配对方法的预测表现高度一致(Pearson's r > 0.9),DeePathNet持续优于其他七种方法(补充图S2A和S2B)。 为验证模型性能,我们在CLP数据集上训练DeePathNet(补充表S1),并通过预测CCLE数据集中的药物反应测试最终模型(补充表S1)。CLP和CCLE重叠的细胞系被排除在测试集之外(参见“材料与方法”)。癌症路径信息以上述方式整合,并以随机森林模型作为基线训练。对所有549种GDSC抗癌药物的测试表现进行了DeePathNet和随机森林的综合总结。DeePathNet在这三种指标上均显著优于随机森林的预测表现(见图3A;P值<0.0001;双尾配对学生t检验;补充表S4)。 为比较DeePathNet与随机森林的预测表现,测量了DeePathNet与随机森林之间的R2差异。其中92%(505/549)的药物为正值,表明DeePathNet在随机森林中的预测表现更优。同样,94%(516/549)药物和76%(415/549)药物分别在MAE和培生r下表现出改善(见图3B)。这表明DeePathNet在大多数抗癌药物中持续优于随机森林的预测表现。DeePathNet中R2改善幅度最大的药物是KIN001-260(图3B),但随机森林预测不佳,导致R2值分布出现长尾(见图3A)。在MAE和Pearson's r中,DeePathNet改善幅度最大的药物是thapsigargin和taselisib(见图3B)。 接着,我们扩展了分析,纳入了来自ProCan-DepMapSanger(44)和CCLE(45)的两个蛋白质组细胞系数据集。ProCan-DepMapSanger 是我们团队最近发布的泛癌蛋白质组数据集,包含949个人类细胞系,补充了CLP的蛋白质组学信息。DeePathNet和随机森林在合并的CLP和ProCan-DepMapSanger 数据集(CLP+;补充表S1),最终模型在扩展后的CCLE数据集上测试,该数据集包含额外的蛋白质组测量(CCLE+;补充表S1)。路径信息如上所述被整合进DeePathNet。DeePathNet在预测549种GDSC抗癌药物时,在这三项指标上均显著优于随机森林检测(图3C;补充表S4)。通过分析每种药物的预测表现,DeePathNet 在大多数抗癌药物中也显著改善了随机森林(Fig. 3D)。DeePathNet改善幅度最大的药物是AZD4877、thapsigargin和BPTES,分别通过R2、MAE和培生r的差值测量(Fig. 3D)。 为了调查DeePathNet预测最准确的药物类型,我们根据549种药物的典型靶细胞通路进行了分组。针对ABL信号和ERK MAPK信号通路的药物,其预测值与实际IC50值之间的平均皮尔逊r数值最高(补充图S3A)。预测最准确的20种药物及其通路详见补充图S3B。 综合这些观察,我们证明DeePathNet通过多项基准分析提升了预测对多种靶向不同信号通路药物反应的预测性能。DeePathNet在药物反应预测方面的表现也通过独立数据集进行了验证。
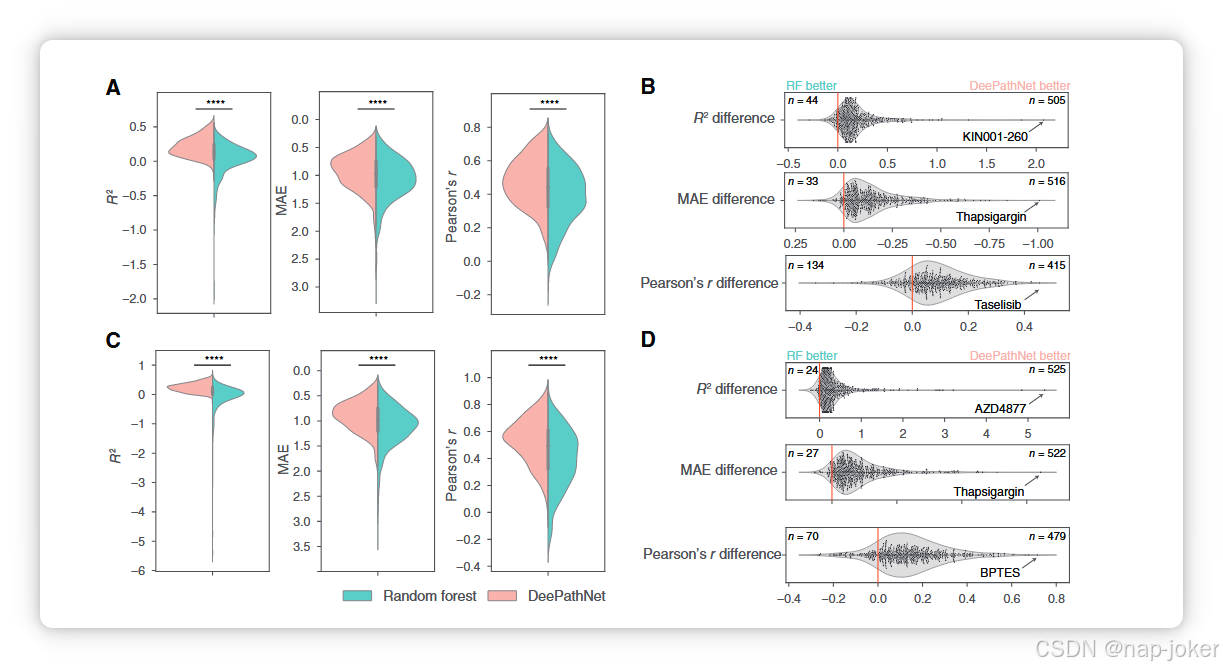
图3 DeePathNet和随机森林在药物反应预测中的推广误差。A小提琴图展示了DeePathNet和随机森林的预测表现,使用CLP作为训练集,并在549种GDSC药物的独立CCLE测试集上评估。MAE的垂直轴是倒置的。,P值<0.0001,由双尾配对学生t检验得出。B、小提琴和群组图显示了每种药物在 DeePathNet 与随机森林之间 R2(上)、MAE(中)和 Pearson's r(下)的性能差异。当药物在R2或皮尔逊r差值为正,或MAE差值为负(MAE水平轴为倒置)时,DeePathNet预测得更准确。DeePathNet或随机森林预测更准确的药物数量分别标注在图的右上角和左上角。每个指标都标注了使用DeePathNet实现最大改进的药物名称。C 和 D,类似于 A 和 B,但使用 CLP+ 作为训练集,CCLE+ 作为独立测试集。
DeePathNet区分癌症分型
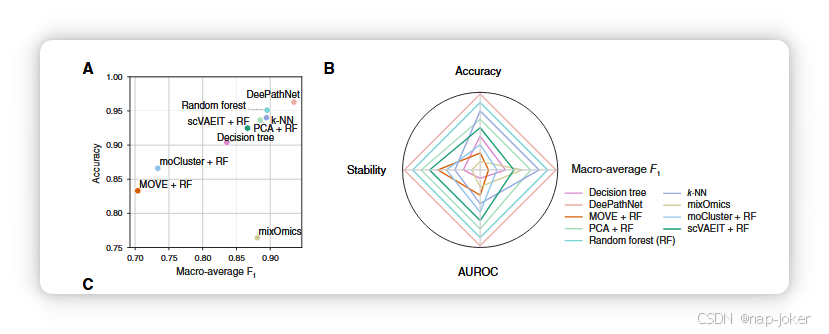
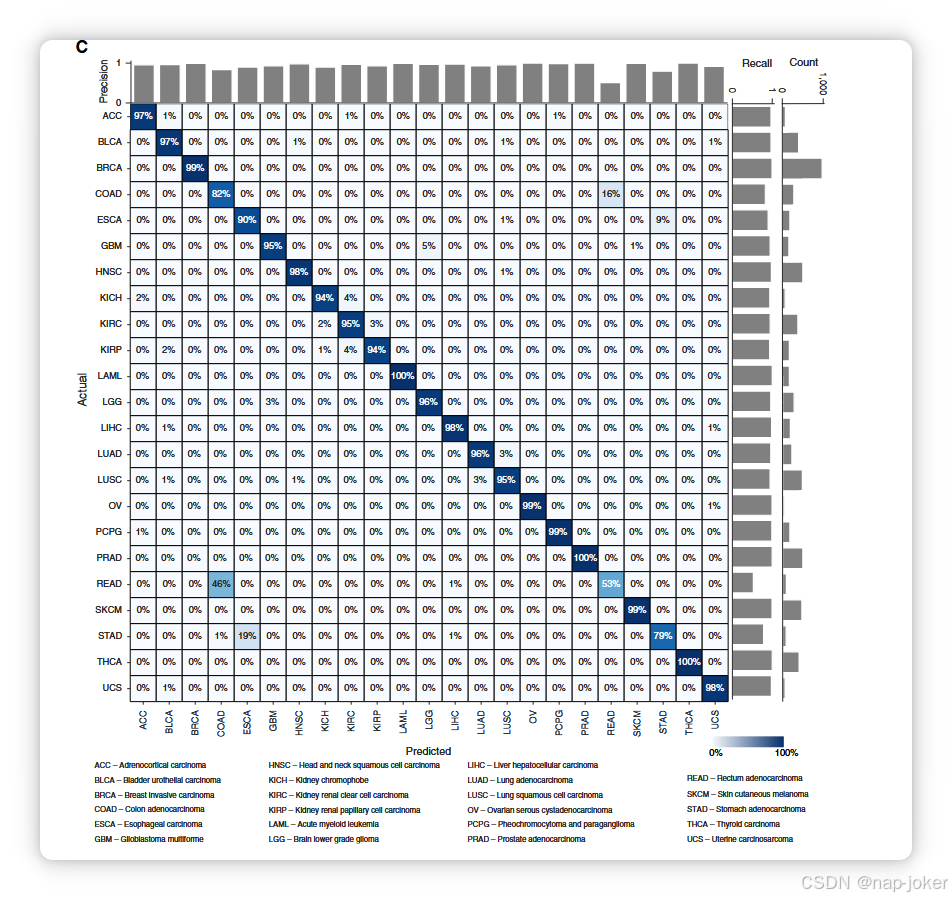
图4 癌症类型分类的性能评估。A,利用TCGA数据集的交叉验证进行模型比较。横轴表示宏观平均F1分数,纵轴表示准确性。B,雷达图,显示模型在四个指标上的排名。更大的封闭面积表示预测性能更佳。C,用于分类23种癌症类型的混淆矩阵。列表示预测标签,行表示实际标签。显示的百分比代表对应癌症类型预测的比例,每行总和为1。对角线代表每种癌症类型的正确预测,百分比表示召回率。条形图显示每种癌症类型的精度(水平轴)、召回率(纵轴,最左侧)和样本数量(纵轴,最右侧)。
为评估DeePathNet的分类任务,我们使用TCGA(52)公开数据对原发性癌症类型进行分类。基因突变、CNV和基因表达特征作为组学数据输入,训练DeePathNet模型将6,356个样本归类为23种癌症类型之一(参见“材料与方法”)。分析中共使用七项指标以确保评估的可靠性。这些指标包括准确率、宏观平均F1评分、精度、召回率(灵敏度)、AUROC、AUPRC和稳定性(参见补充材料和方法)。LCPathways的整合方式与药物反应预测相同。在基准测试中,弹性网被k-NN(53)取代。之所以这样做,是因为弹性净正则化在癌症类型分类中较少使用,而k-NN则是更广泛采用的分类方法。在所有九种方法中,特征整合和超参数设置均与药物反应预测相同。 由于缺乏涵盖23种癌症类型的独立数据集,对这九种方法在TCGA数据集上进行了交叉验证,评估指标的平均值和95%置信区间被报告为泛化误差的估计值。DeePathNet在准确率和宏观平均F1得分上持续优于其他八种机器学习方法(见图4A;补充表S5)。相比之下,其他方法如混合经济学仅在某一指标上表现良好,表明这些方法可能适用于某些场景,但无法很好地泛化到不同预测任务(见图4A)。使用包括准确率、宏观平均F1分数、AUROC和稳定性在内的四项指标评估每种方法的性能,显示DeePathNet始终位居首位,随机森林排名其次(图4B;补充表S5)。 为了进一步研究DeePathNet对每种癌症类型的表现,采用混淆矩阵可视化每个样本的预测和实际癌症类型,并标注样本数量、精度和召回率(见图4C)。DeePathNet对大多数癌症类型实现了超过0.95的召回,其中急性骨髓性白血病、胰腺腺癌和甲状腺癌是分类最准确的前三大癌症类型。直肠腺癌是回忆率最低的癌症类型,46%的样本被错误归类为结肠腺癌。后者的结果并不令人意外,因为结肠和直肠是相邻的组织类型,具有高度相似的特征,这两种癌症类型常被归为一类(54),并采用相似的化疗方案(54)。召回率第二低的癌症类型是胃腺癌,19%的胃腺癌样本被错误归类为食管癌。这可以通过它们相似的组织病理学以及胃腺癌和食管癌的解剖学接近来解释(55)。接着,AUROC和AUPRC针对每种癌症类型进行了检查,均在所有癌症类型中表现出较高表现,惟直肠腺癌的AUPRC除外,因为直肠腺癌与结肠腺癌的组织距离较近(补充图S4A和S4B)。
DeePathNet区分乳腺癌亚型
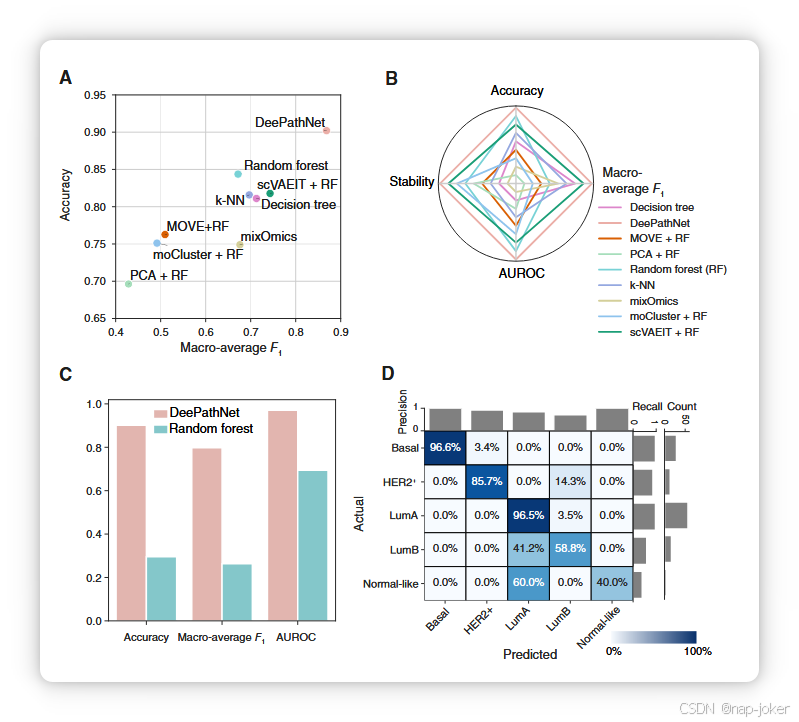
图5 乳腺癌亚型分类的性能评估。A,通过交叉验证进行模型评估。横轴表示宏观平均F1分数,纵轴表示准确率。B,雷达图,显示模型在四个指标上的排名。更大的封闭面积代表更好的分类性能。C, 性能指标显示在使用CPTAC数据作为独立验证测试集时,DeePathNet和随机森林的泛化误差。D,使用CPTAC数据作为独立验证测试集时的泛化误差的混淆矩阵。统计量的注释方式与图4C描述相同。
基因突变、CNV和基因表达特征被用于训练DeePathNet模型,用于根据以下PAM50测试(56)条件对五种乳腺癌亚型(腔内A型、腔内型B型、HER2+、基础型和正常型)进行分类。共使用了TCGA数据集中的974个乳腺癌样本进行训练,并纳入了CPTAC中122个乳腺癌样本的队列作为独立数据集,以评估泛化误差。 首次对所有九种方法进行交叉验证,报告评估指标的平均值和95%置信区间,作为泛化误差的估计值。DeePathNet在准确率和宏观平均F1分数方面相比其他方法有显著提升(见图5A;补充表S6)。AUROC的表现提升相对较小,但具有统计学显著性(学生t测试P值<5 10 4;补充表S6)。随后,这些方法根据与癌症类型分类相同的四项指标进行排名。DeePathNet在四个指标中均表现最佳,随机森林排名第二(图5B)。其他方法显示不同指标的绩效排名不一致,表明综合评估需要使用多个评估指标。 为了验证模型在独立测试集上的表现,DeePathNet模型在TCGA乳腺癌队列上进行了训练,最终模型则在独立的CPTAC乳腺癌队列上测试。以随机森林为基准测试,DeePathNet在独立测试集上的泛化误差要低得多(图5C;补充表S7)。接下来,通过混淆矩阵评估了DeePathNet的每个子类型的推广误差。DeePathNet在分类基础亚型方面实现了最高的精度和召回率(96.6%;图5D),该亚型大多数肿瘤为高级别且预后较差(57)。最难分类的亚型是正常样,其中五个正常样样本中有三个被错误归类为luminal A(见图5D)。腔型A型和正常样亚型传统上难以区分,因为它们共享相同的IHC标志(57)。正常样亚型在临床中使用较少(58)。AUROC(补充图S5A)和AUPRC(补充图S5B)的进一步分析显示,DeePathNet在每个亚型的预测表现都很高。总体而言,这些结果展示了DeePathNet在癌症亚型分类方面通过交叉验证和独立数据集验证的表现。
DeePathNet提供模型的可解释性
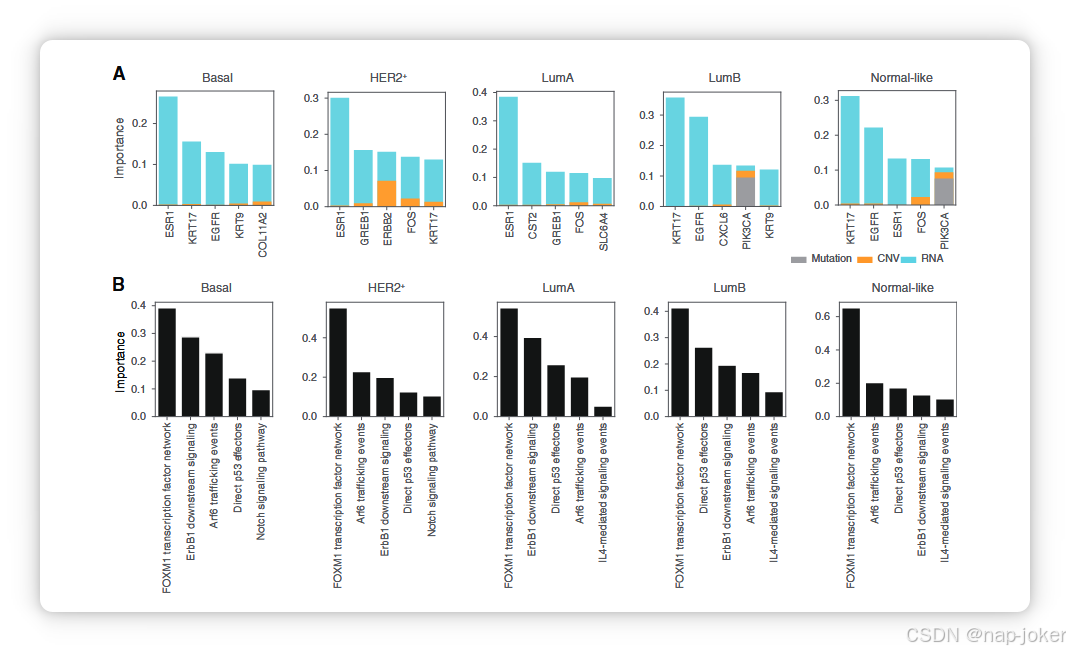
图6 DeePathNet模型按组学层级和通路层面特征重要性进行解释。A,堆叠条形图,显示每种组学数据类型中前五基因的组学级特征重要性(用灰色、黄色和蓝色表示)。B,显示DeePathNet路径水平的条形图显示前五条路径的重要性。
DeePathNet模型可通过利用SHapley加性解释(SHAP;参考文献59)和层级相关性传播(LRP;参考文献60)的特征重要性,在组学和通路层级进行解释。SHAP将预测归属于所有特征,并为每个特征分配重要性值。同时,LRP假设分类器可以分解为多个计算层,这些层是特征提取的一部分。因此,SHAP和LRP是事后模型解释方法,在DeePathNet训练完成后,建立特征值与预测之间的关系。采用乳腺癌亚型分类来证明模型解释。 为了在组学层面解释模型,使用了SHAP计算特征重要性。具体来说,对前五基因的特征重要性进行了计算和可视化,形成包含每个乳腺亚型的组体数据类型的堆栈条形图(见图6A)。DeePathNet能够识别已知生物标志物基因作为主要特征,如ESR1、ERBB2和KRT17,其基因表达常被用于临床中确定PAM50亚型(图6A;参考文献56)。大多数基因的高度特征重要性归因于转录组数据(图6A),这与PAM50分类为基于RNA的亚型一致(56)。 为了在通路层面解释模型,使用了LRP来计算特征重要性。由于癌症通路以编码向量形式表示,汇总多组学信息,癌症通路的特征重要性是联合计算所有组学数据类型的。对于每个癌症亚型,排名中特征重要性值最高的前五条通路(见图6B)。DeePathNet确定FOXM1转录因子网络是预测所有PAM50亚型最重要的通路(见图6B)。FOXM1在不同乳腺癌亚型中表现出明显的表达模式,被视为乳腺癌治疗中有前景的候选靶点(61)。FOXM1也是管腔A型和B型的不良预后因子(62)。研究显示,ARF6通路在三阴性乳腺癌中表达过度,并与乳腺癌侵袭和转移相关(63)。同样,Notch信号通路参与细胞增殖、凋亡、缺氧及上皮向间充质的转变,并在HER2阳性和三阴性乳腺癌中被发现表达过度(64)。 综合来看,这些发现表明DeePathNet通过在组学层面和途径层面都提供了特征重要性,提供了可靠的模型解释和坚实的生物学基础。
消融研究
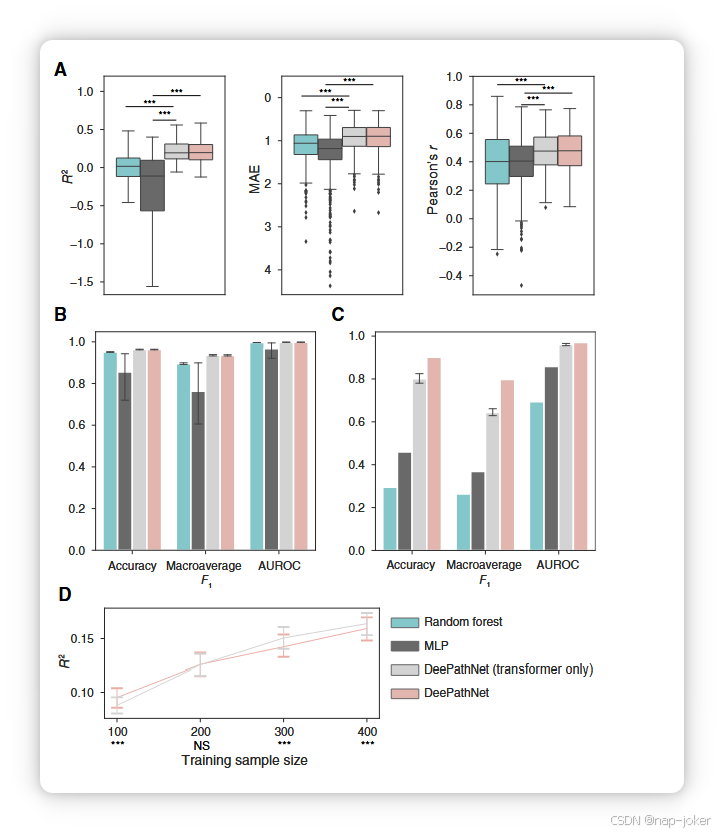
图7 使用仅变换器模型和纯神经网络(MLP)对DeePathNet进行消融研究。A,箱形图显示了549种药物中随机森林(绿色)、MLP(深灰色)、带随机布线通路的DeePathNet(浅灰色)和DeePathNet(粉色)的独立测试测试的预测表现。R2的离群值被隐藏,以便更清晰地可视化。B,条形图展示了四个模型在癌症类型分类交叉验证中的预测表现。C,Bar图显示乳腺癌亚型分类独立检验集的预测表现。置信区间代表平均值的95%置信区间(n1/4个实验)。D,下采样分析显示,DeePathNet在较小训练样本量下,在药物反应预测方面表现优于随机有线通路的DeePathNet。实线表示平均R²,置信区间代表平均值的95%置信区间(n1/4个实验)。,P < 0.001,配对t检验。
最后,我们通过分别检查 DeePathNet 的两个主要组件:路径编码器和变压器模块,评估了其预测能力。为了评估通过整合癌症通路知识带来的额外预测强度,我们通过随机修改DeePathNet的通路定义,设计了一个对照组。这个过程重复了十倍。与仅变压器模型相比,DeePathNet在乳腺癌亚型分类的所有指标上提供了更准确的预测,而药物反应预测和癌症类型分类也观察到类似的结果(见图7A–C;补充表S8和S9)。此前也观察到随机配置与手动设计神经网络之间的性能相当(26, 65)。在不失去预测能力的前提下,利用人类知识设计的神经网络被发现能提供更有意义的模型解释,从而可能带来生物学发现(26, 65)。进一步研究复制了前一研究中观察到的现象(65),证明利用已知生物学知识构建的神经网络在样本量有限时优于仅变压器模型(图7D)。接下来,我们用MLP作为一个不使用通路信息或变压器的普通神经网络对DeePathNet进行了基准测试。当同时配备通路信息和变压器模块时,DeePathNet在所有考虑的指标和任务中实现了显著更准确的预测(图7A–C;补充表S8和S9)。
总之,无论是否存在路径知识,DeePathNet与变压器模块集成时,都比普通神经网络和随机森林提供了显著更高的预测能力。纳入通路知识提升了DeePathNet在乳腺癌亚型分类中的准确性,并促进了通路层模型的解释。
讨论
DeePathNet通过整合癌症通路,拓宽了变换器在癌症多组数据集成中的应用,成功克服了大多数现有机器学习方法的局限性,这些方法不考虑已知的癌症生物学。DeePathNet将多组数据与癌症通路知识相结合,准确预测药物反应并分类癌症类型及亚型。变换器模块的自我关注机制动态模拟了通路间的相互依赖关系,从而捕捉了不同生物过程之间的调控效应以及失调的影响。与传统机器学习模型中每个输入特征的影响在训练后固定不同,自注意机制使模型能够在推断过程中为每个样本的不同路径分配不同重要性。这种动态权重允许对通路交互进行更细致的建模,针对每个输入样本的具体情境量身定制。通过直接建模路径而非多组特征,DeePathNet 还缓解了多组数据分析中常见的维度诅咒(48)。 DeePathNet的预测性能通过一项回归和两项分类任务进行了评估和验证。评估规模大于以往类似研究(8,11),使用多个大型数据集和多种指标,结合交叉验证和独立测试。基于变换器架构,DeePathNet 优于那些不包含路径信息、仅依赖 omic 数据获取输入特征的其他机器学习方法。在独立数据集上验证DeePathNet模型时,低泛化误差表明即使这些独立数据集中实施了不同的实验方案,DeePathNet依然良好。DeePathNet在通路层面提供模型解释,目前尚无其他多组集成工具能够预测药物反应或分类癌症类型及亚型。DeePathNet能够突出显示已知的生物标志物,用于预测乳腺癌亚型,包括ESR1、ERBB2和FOXM1网络通路。这表明其他排名更高的特征,如ERBB1,下游信号通路,可能为癌症生物学和药物发现提供新颖见解。由EGFR激活的ERBB1下游信号通路调控细胞生长、存活和分化等关键过程,进一步研究可能揭示促成药物耐药的新治疗靶点或机制。 在我们的消融研究中,我们观察到,尽管仅变压器模型在预测表现上与DeePathNet相似,但通路信息的加入带来了显著优势。具体来说,变换器模块通过其自注意机制有效模拟特征间的复杂关系,提高了准确性。通过整合通路信息,DeePathNet不仅保持了高水平的预测性能,还赋予通路层面特征的重要性,使我们能够识别与预测结局相关的最重要的癌症通路。此外,在数据稀缺的环境中,结合先前的生物学知识尤其有益,可以指导模型并提升准确性。相比之下,在数据丰富的环境中,模型可以独立学习这些关系,从而减少预定义路径知识对预测表现的直接影响。 尽管进行了全面评估,DeePathNet目前仅设计支持基于基因的功能。其他组学数据类型,如磷酸酶组学、代谢组学和图像,不能直接作为通路编码器的输入。因此,DeePathNet 未来的工作包括扩展模型以支持任何数据模态,这可以通过使用不同类型的编码器将特征映射到通路空间来实现。此外,对每个基准测试方法进行更全面的超参数调优,可以提供更可靠的比较,并有可能提升它们相较于DeePathNet的性能。乳腺癌亚型分类中展示的可解释性分析可以扩展到其他预测任务,如药物反应预测和癌症类型分类,进一步展示DeePathNet的可解释性及其在多样应用中揭示有意义生物学见解的潜力。与此同时,随着大量蛋白质组和代谢组数据集的日益普及,DeePathNet的预测能力将提升,因为深度学习将随着数据增加而获得性能提升(66)。 总之,DeePathNet结合了多组学、深度学习和现有生物学知识,通过模型解释准确预测癌症表型。DeePathNet的应用有望带来更准确的诊断和预后,并帮助研究人员理解未知的癌症机制并优先确定潜在药物靶点。

AtomGit 是由开放原子开源基金会联合 CSDN 等生态伙伴共同推出的新一代开源与人工智能协作平台。平台坚持“开放、中立、公益”的理念,把代码托管、模型共享、数据集托管、智能体开发体验和算力服务整合在一起,为开发者提供从开发、训练到部署的一站式体验。
更多推荐

 13
13 0
0- 0



 已为社区贡献27条内容
已为社区贡献27条内容

所有评论(0)