论文学习——通过蛋白质片段-环境比对实现自我监督口袋预训练
论文题目:SELF-SUPERVISED POCKET PRETRAINING VIA PROTEIN FRAGMENT-SURROUNDINGS ALIGNMENT
通过蛋白质片段-环境比对实现自我监督口袋预训练(Bowen Gao1∗, Yinjun Jia2∗, Yuanle Mo3, Yuyan Ni4, Weiying Ma1, Zhiming Ma4, Yanyan Lan1,5†)Published as a conference paper at ICLR 2024
个人总结:
这个文章提出一个方法叫ProFSA 主要是用来解决 药物研发中,蛋白质和小分子结合数据太少的问题。
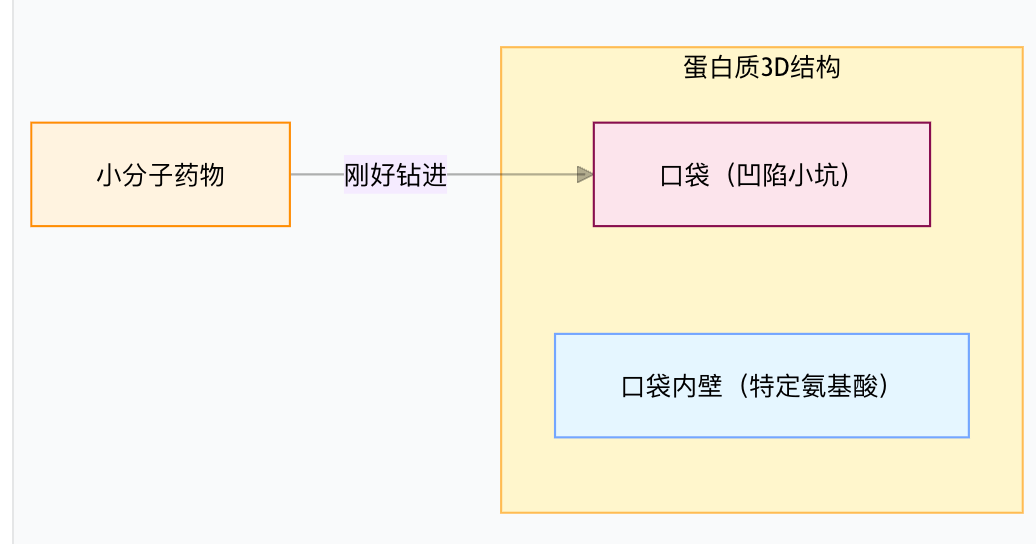
主要通过从蛋白质切下一块片段当做假药物,构造了500万组假药物-蛋白质口袋的数据,训练出一个ai模型,去判断口袋能不能放得下药物,或者能不能结合的牢靠。 就类似蛋白质口袋是 “钥匙孔”,小分子是 “钥匙”,只有形状、材质匹配的钥匙,才能插进钥匙孔并发挥作用。
具体细节:
步骤1:从蛋白质上切1-8个氨基酸组成的片段,把这个氨基酸片段当做假小分子(因为有相似的化学结构,同时能与口袋结合)
步骤2:每个假小分子周围,一定会存在凹陷区域,把他叫做假口袋,以片段为中心,找周围 6 埃(原子尺度的距离单位)内的蛋白质残基,这些残基构成的凹陷就是 “假口袋”;同时要做一定的修正工。给片段的两端加化学基团,让它更像真实小分子。
步骤3:造出500多万种组合,并且保证与真实数据中的口袋大小结合程度一致。
步骤4:让模型学习正确的配对形式和不正确的配对形式,从而让他找到规律扩展到真实的情况中。
借鉴到酶优化中?
能否在目标酶的结构中也切,然后学习。
或者是在编码的时候考虑3D结构特征,在对某一个节点进行突变的时候,同时考虑到突变以后跟口袋是否还匹配?
摘要
- 口袋表示(poket representations play)在多种生物医学应用中发挥着重要作用,如可药性估计、配体亲和力预测和新药物设计。虽然现有的几何特征和预训练表示已显示出有前景的结果,但它们通常处理独立于配体的口袋,忽视了配体之间的基本相互作用。
- 然而,PDB数据库中有限的口袋配体复杂结构(不到10万对非冗余对)阻碍了大规模的交互建模预训练工作。
- 为解决这一限制,我们提出了一种新型口袋预训练方法,利用高分辨率原子蛋白结构的知识,辅以高效的预训练小分子表示。通过将蛋白质结构分割为类药物片段及其相应的口袋,我们获得了配体-受体相互作用的合理模拟,产生了超过500万个复合物。随后,口袋编码器会以对比方式训练,以与某些预训练小分子编码器提供的伪配体表示对齐。我们的方法名为ProFSA,在包括口袋药物可药性预测、口袋匹配和配体结合亲和力预测等多个任务中实现了最先进的性能。
- 此外,我们的工作为利用高质量且多样化的蛋白质结构数据库,为缓解蛋白质-配体复杂数据稀缺开辟了新途径。代码和数据可在 https://github.com/bowen-gao/ProFSA 获取。
引言
在药物发现方面,人工智能驱动的方法取得了显著进展,越来越多地聚焦于蛋白质口袋(蛋白质口袋是蛋白质 3D 结构上的 “功能小坑”,由折叠形成,通过形状、氨基酸组成决定能结合的分子)的表现。这些口袋在通过疏水力、氢键、π堆叠和盐桥等多种相互作用结合小分子配体中发挥关键作用。而传统的手工特征如曲率、溶剂可接触的表面积、水病和静电学提供了宝贵的领域洞见,其计算需求和无法捕捉复杂交互限制了其实用性。
相比之下,数据驱动的深度学习方法,如Gao等(2022)、Karimi等(2019)和̈Oztu ̈rk等(2018)提出的双塔架构,展现出潜力,但受限于复杂结构数据的稀缺(BioLip2数据库中不到10万对非冗余数据(Zhang等,2023))。为解决这一限制,自监督学习技术,如Zhou等人(2023)提出的,重点是恢复损坏数据,使其能够利用大量无界口袋数据进行表示学习。然而,由于配体侧缺乏适当的监督信号,这些方法常常忽视了重要的配体-口袋相互作用。为了充分发挥口袋表示学习的潜力并强调口袋与配体之间的相互作用,迫切需要开发大规模数据集和创新的预训练方法。
提出的方法
利用蛋白质片段-环境比对技术进行大规模数据构建和口袋表示学习。该方法的基本原则是认识到蛋白质配体相互作用遵循与蛋白质间相互作用相同的物理原理,因为它们共享有机官能基,如苯丙氨酸中的苯基和谷氨酸中的羧基。因此,我们可以通过从蛋白链中提取类药物片段来模拟蛋白质-配体相互作用,并在预训练的小分子编码器指导下学习配体感知口袋表示,从而策划出更大、更多样化的数据集。这些编码器已被许多先前研究广泛探索,使用大型小分子数据集,例如 Uni-Mol(Zhou 等, 2023年),Frad(Feng等,2023年)
为了弥合片段口袋对和配体口袋对之间的差距,我们采用了三种策略。首先,我们关注具有长程相互作用的残基,排除那些受真实配体-蛋白质复合物中缺失的肽键约束的残基(Wang 等,2022a)。其次,我们根据片段-口袋对的相对埋藏表面积(rBSA)和口袋与配体大小的联合分布进行采样,以模拟真实配体。第三,我们通过应用终端修正(Marino 等,2015;Arbour 等,2020)到片段。这导致PDB数据库中总共计550万对对。
利用该数据集,我们引入了一种分子引导对比学习方法,以获得配体感知口袋表示。我们的方法训练口袋编码器区分同批次中的正配体和其他负样本。我们的对比框架能够将多样的相互作用模式编码到口袋表示中,无论蛋白质片段与真实配体之间是否不一致。为抵消用作配体的肽片段带来的偏差,我们将口袋编码器与预训练的小分子编码器比对,其权重固定。该预训练编码器作为指导,将大量分子数据集中的结合特异性和生物学相关信息传输到我们的口袋编码器。
文章的主要贡献如下:
1. 提出一种新颖的可扩展成对数据合成流程,通过从仅蛋白质数据中提取伪配体口袋对,有望应用于AlphaFold(Jumper等,2021)或ESMFold(Lin等,2023)生成的更大预测结构数据。
2. 引入一种新的分子引导片段环境对比学习方法用于口袋表示,该方法自然地提炼出预训练小分子编码器的全面结构和化学知识。
3. 在多个下游应用中实现了显著的性能提升,包括仅需口袋数据的任务以及涉及口袋配体对数据的任务,凸显了ProFSA作为药物发现领域强大工具的潜力。
背景及相关工作
A.口袋预训练数据
目前可用的蛋白口袋数据均来自蛋白质数据库(PDB))。最著名的数据库是PDBBind最新版本(v2020)包含19,443对蛋白-配体。虽然PDBBind数据库关注具有亲和力数据的蛋白配体配对,但其他数据库也收集无亲和力配体配对。Biolip2是最全面的数据库之一,包含467,808对口袋配体配体,但只有71,178对是非冗余配体(2023-09-21的周版)。由于结构生物学实验成本高且耗时,短期内这类数据库难以实现显著增长。另一方面,个人可能依赖口袋预测软件,如Fpocket(Zhou等,2023)来扩展口袋数据集。然而,这些程序识别的口袋本质上缺乏配体,因此对蛋白质-配体相互作用的了解不足。
B.口袋预训练方法
近年来,多种预训练方法在创建蛋白质口袋的有效表示方面表现出卓越的性能,这些表示可用于多种下游任务。一些方法将结合位点视为蛋白质靶点的一部分,直接对蛋白质结构进行预训练或序列。其他方法则作用于从靶点分离的显式口袋,使模型更具聚焦性。其中一种方法是 Uni-Mol(Zhou 等,2023),它提供了一个通用的三维分子预训练框架,涉及一个基于300万个口袋数据进行预训练的口袋模型。然而,该方法缺乏对口袋与配体之间相互作用的具体关注。此外,CoSP(Gao等,2022)采用协监督的预训练框架,同时学习口袋和配体表示。然而,由于数据不足导致化学空间缺乏多样性,所学表示并不完全令人满意。
C.分子预训练方法
分子表征学习极大地促进了分子性质预测和药物-靶点相互作用预测等任务中的药物发现。继自监督学习在自然语言处理和计算机视觉中取得巨大成功后,各种预训练方法被开发出来,以解决该领域标记数据的稀缺性。最初,大多数方法在一维 SMILES 字符串(Wang 等,2019)或二维图上运行,并采用掩蔽语言模型、自我对比目标或一维与二维表示之间的对齐。然而,鉴于分子三维几何结构在决定其物理和化学性质中的重要性,近期方法越来越多地聚焦于利用三维分子数据进行预训练,为分子结构提供了更全面的理解。
本文的方法
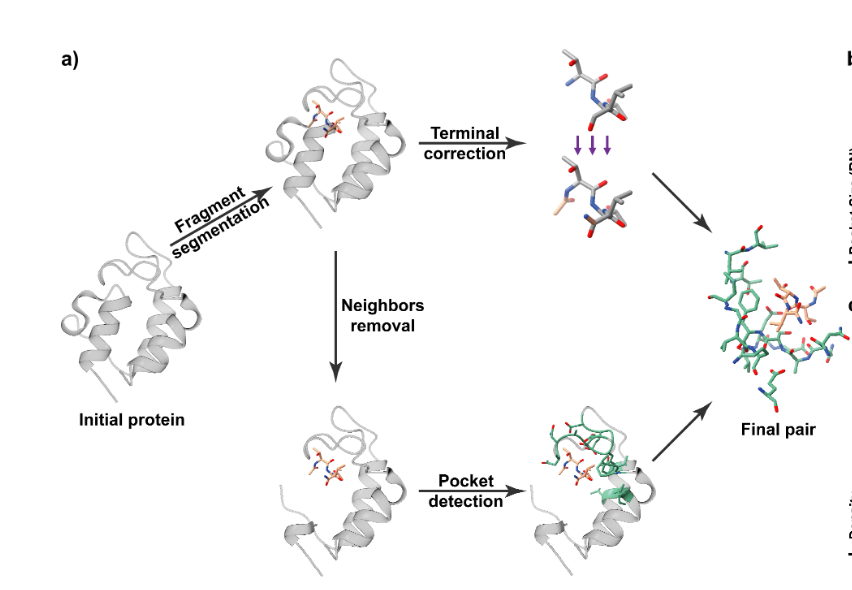
A.从蛋白质数据构建伪配体口袋复合物
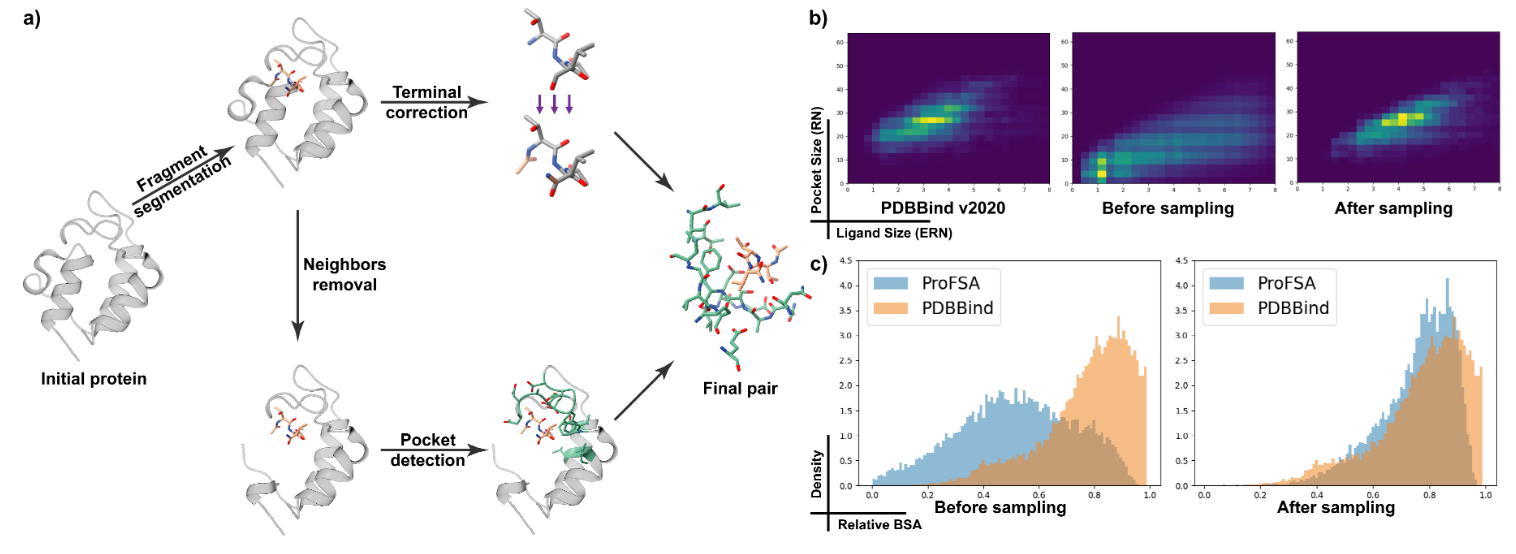
图1:a)从蛋白质中分离口袋配体对的流程;b)PDBBind数据集中口袋大小与配体大小的联合分布,分别为分层抽样前和分层抽样后的ProFSA数据集;c)ProFSA数据集与PDBBind数据集在配体-口袋对rBSA分布方面的比较。
为了解决实验确定的口袋配体对的稀缺性问题,我们开发了一种创新策略,在PDB存储库中挖掘大量仅蛋白质的数据。我们推断,关于非共价相互作用的知识可以从蛋白质推广到小分子。因此,我们从一个与配体极为相似的蛋白质结构中提取片段,并将周围环境指定为该伪配体的对应囊袋。
我们的数据构建过程从非冗余的PDB条目开始,这些条目聚集在70%的序列同性阈值处。该过程包括两个阶段:伪配体构建和口袋构建,如图1所示。第一阶段,我们迭代分离从N端到C端的片段,从1到8个残基,避免任何不连续位点或非标准氨基酸。为了解决片段分割过程中肽键断裂带来的偏差,我们对末端修正,特别是乙酰化和酰胺化,分别对N端和C端进行,最终形成伪配体。第二阶段,我们排除获得片段两侧最近的五个残基,重点关注长程相互作用。然后,我们将与片段相距6 ̊A范围内、包含至少一个重原子的周围残基(按照Uni-Mol的设置,其他截断值见表10)指定为口袋。该过程产生了成对的最终口袋和伪配体。
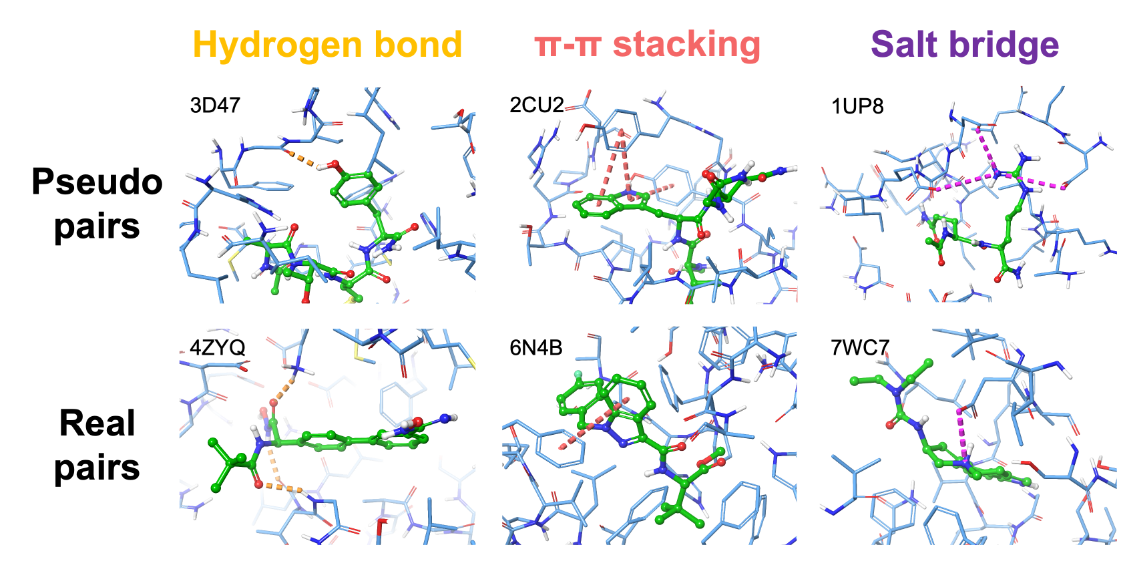
通过上述程序生成的配体口袋对可以模拟大多数非共价相互作用。如图2所示,氢键形成于蛋白质主链和极性氨基酸如丝氨酸或组氨酸之间;芳香族氨基酸(如酪氨酸、色氨酸和苯丙氨酸)之间常见π−π堆叠;此外,带相反电荷的氨基酸如精氨酸和谷氨酸之间也存在盐桥。图中未包含一些其他罕见的相互作用类型,但它们也存在于生成的数据集中。例如,带正电的精氨酸和苯丙氨酸之间可能存在阴离子π相互作用;甚至还有一些非典型的相互作用,如C-H··π已被报道(Brandl 等,2001)。因此,正如Polizzi和DeGrado(2020)观察到的,蛋白质内相互作用可以很好地模拟蛋白质-配体相互作用。
对伪复物进行采样,以近似PDBBind数据集的分布。所得数据集在rBSA和口袋配体大小联合分布方面与PDBBind相似,如图1b和图c所示。
B.口袋碎片空间中的对比学习
我们提出了一种在蛋白质-片段空间中的对比学习方法,以将交互知识注入口袋表示。如先前所示(Wang 和 Isola,2020),从正向数据中实现特征比对的能力是对比学习的重要特征。先前研究表明,对比学习在图像-文本(Radford 等,2021)和口袋-配体相互作用(Gao 等,2024)中都有效。具体来说,对于蛋白质口袋p及其对应的伪配体l,我们将其归一化编码载体表示为s和t。设gT和gS分别表示配体和蛋白质预测网络。然后使用两种不同的对比损失来促进训练过程:
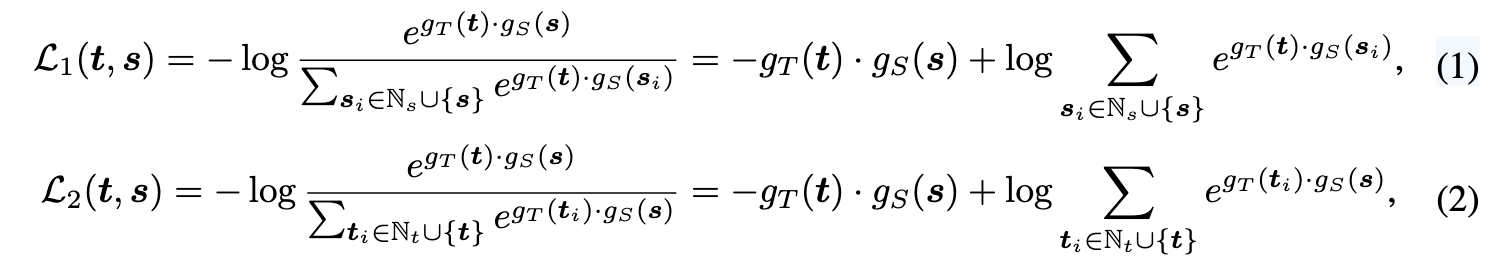
其中Ns和Nt分别指的是蛋白质口袋和伪配体的批次阴性样本。第一个损失的主要目的是识别给定伪配体时,批次样本中的真实蛋白口袋。同样,第二个损失旨在识别给定口袋对应的配体片段。
因此,我们训练过程中使用的最终损失为:
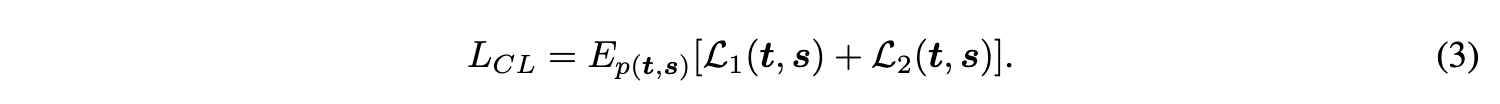
我们的方法区别于传统的对比学习方法,保持分子编码器的参数不变(见图3,左侧面板)。尽管我们生成的数据集与PDBBind数据集高度对齐,但值得注意的是,伪配体在化学性质(如logP和旋转键数)方面仍与真实配体不同(图9)。固定分子编码器可以缓解真实配体与伪配体之间的差异,使我们能够从大型预训练分子编码器中提取所学的生物化学知识,并进一步降低计算成本。因此,与固定片段表示对齐的口袋表示也编码了具有生物学意义的属性(图3,右侧面板)。固定编码器优势的实证证据见表5,相应定理和严谨证明见附录B。
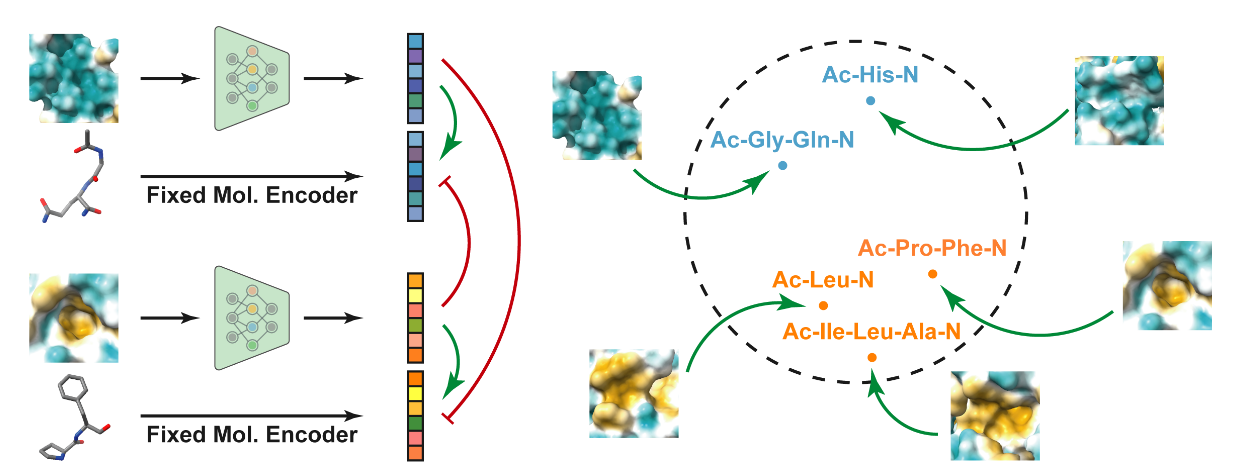
图3:蛋白质片段-环境比对框架的示意。口袋编码器通过我们的口袋编码器编码,该编码器训练为与固定预训练分子编码器给出的片段表示对齐。一个简化的水病相关示例(蓝色或橙色表示)说明了预训练分子编码器识别的片段属性可以指导口袋表示学习。

AtomGit 是由开放原子开源基金会联合 CSDN 等生态伙伴共同推出的新一代开源与人工智能协作平台。平台坚持“开放、中立、公益”的理念,把代码托管、模型共享、数据集托管、智能体开发体验和算力服务整合在一起,为开发者提供从开发、训练到部署的一站式体验。
更多推荐

 9
9 0
0- 0



 已为社区贡献1条内容
已为社区贡献1条内容

所有评论(0)