
R统计-微生物群落结构差异分析及结果解读
前一段时间忙着转录组数据分析,说好的群落结构差异分析结果解读,严重拖更。期间微生信生物公众号发布了微生物群落分析的详细流程。感觉写的详细,跟我这种已经转了1年半行的确实不一样。虽然.......
前一段时间忙着转录组数据分析,说好的群落结构差异分析结果解读,严重拖更。期间微生信生物公众号发布了微生物群落分析的详细流程。感觉写的详细,跟我这种已经转了1年半行的确实不一样。虽然......,但是我还是想把硕士期间的笔记整理出来,就当是青春的记录了。大家想看更全面详细的分析流程,可以看推荐阅读
1. 群落结构差异分析
为了防止大家已经忘记了数据格式和运行代码,这里还是先把分析重新跑一遍。我一般使用Rmarkdown记录代码和运行结果,所以路径设置会有一些不一样,大家可以创建一个Rmarkdown运行,也可以直接使用setwd()设置路径。
# 1. 设置工作路径,老生常谈,设置工作目录和查看目录内容一定是第一步
knitr::opts_chunk$set(echo = TRUE)
knitr::opts_knit$set(root.dir="D:\\2018_04_23")
getwd() # 查看工作目录
dir() # 查看工作目录内容
# 2. 调用R包
install.packages("ecodist")
library(vegan)
browseVignettes("vegan") # 查看R包的帮助文件
library(ecodist)
# 3. 数据读入
spe=read.csv("spe.csv",header=T,row.names=1,sep=",", colClasses = c("character",rep("numeric",times=8)) )# 群落组成数据,物种组成数据是整数,为了使读入数据格式为数值型,设置colClasses,8为物种种类。
# ?rep # 查看函数用法和函数中参数意义
spe
group=read.csv("group.csv",header=T,row.names=1,sep=",")#分类数据,包含两种类别,grazing和soil depth
group
env=read.csv("env.csv",header=T,row.names=1,sep=",") #环境数据:土壤理化因子
env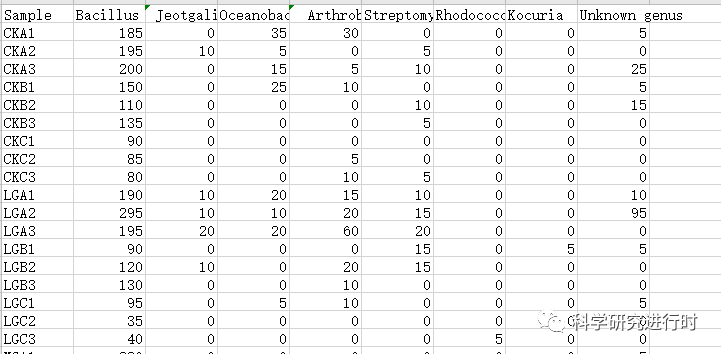
图1|微生物群落物种组成数据。可以对物种数据进行标准化,也可以使用DESeq2包对物种组成数据进行转化(DESeq主要用于转录组数据分析,功能也可以用于微生物分析)。
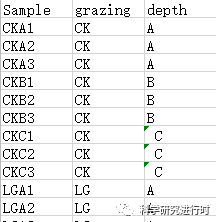
图2|分类变量。
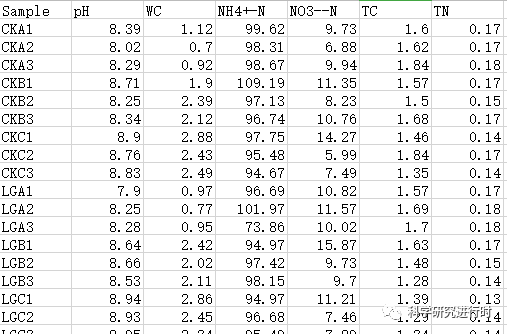
图3|环境因子数据。可以对数据进行转换以排除环境因子本身数值大小造成的误差(log(x)、log(x+1)等,根据数据情况选择)。
2. ADONIS/PERMANOVA-PCoA
PERMANOVA(ADONIS)的中文名称为置换多元方差分析或非参数多元方差分析,利用各种组间距离指数对总方差进行分解,可以分析不同分类因子对群落差异的解释度,并使用置换检验进行统计学检验。参数检验要求分析数据是正态分布和具有方差齐性,非参数检验则对样本数、数据分布和方差齐性要求不高,因此应用可以比较广泛。
2.1 分析流程
群落结构差异分析中使用的β-多样性指数可以根据数据情况进行选择,一般物种数据选择bray-curits或者jaccard等指数。
# PERMANOVA群落结构差异分析-选择bray-curits index
grazing.bray = adonis(spe ~ grazing,data = group,permutations = 999,method="bray") # 用grazing分组spe,并进行Adonis分析,分析结果赋予grazing.bray
grazing.bray # 只能看分类因子整体上是否有差异
grazing.bray$aov.tab
grazing.bray$coefficients
grazing.bray$coef.sites
## 双因素交互影响
NR.bray=adonis(spe ~ depth*grazing,data = group,permutations = 999,method="bray")
NR.bray
# 多重比较-放牧分类因子中包括CK、LG、MG和HG分类,对它们进行两两比较
source("pairs_test.r") # 导入多重比较函数,在上篇推文“R统计-微生物群落结构差异统计分析”中有写,这里为节省篇幅,直接用source()导入
diff = pairwise.adonis(spe, group$grazing, sim.method="bray", p.adjust.m= "bonferroni") # 多重比较,也可使用RVAideMemoire包进行,使用方法推荐阅读微生信生物的文章
# 输出数据
sink(file = "PERMANOVA-bray-curits.csv",append = T) # append=T表示允许向文档中追加内容
grazing.bray
NR.bray
sink() # 将PERMANOVA$call的结果都输出到PERMANOVA-bray-curits.csv文件中
## 将多重比较数据输出到同一文档中
write.table(diff,file="PERMANOVA-bray-curits.csv",append=T,sep = "\t",quote = F,row.names=F) # quote = F,表示不输出字符"";row.names=F,表示不输出行名* β-多样性:沿环境梯度不同生境群落之间物种组成的的相异性或物种沿环境梯度的更替速率也被称为生境间的多样性(between-habitat diversity)。
2.2 分析结果解读
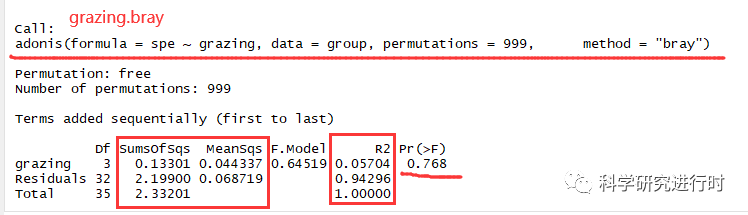
图4|PERMANOVA grazing.bray$aov.tab结果。
* grazing,分类因子
* Df,自由度
* Sumsofsqs,总方差(离差平方和)
* MeanSqs,均方差,=Sumsofsqs/Df
* F.model,F检验值
* R2,不同分类对组间群落差异的解释度,=分类方差/Total方差,其值越大表示分类因子对差异的解释度越大。ps. 其实这里的的解释度,我一直认为可以把它当作一种相关性。
* Pr, P检验值,p>0.05表明基于放牧分类因子的群落结构差异不具统计学意义
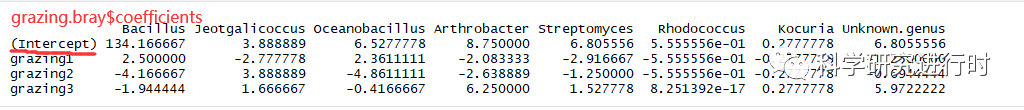
图5|grazing.bray$coefficients结果。
* coefficients,放牧分类因子与群落中每个物种的相关性
* Intercept,截距

图6|grazing.bray$coef..sites 结果。
* grazing与每个样本的相关系数
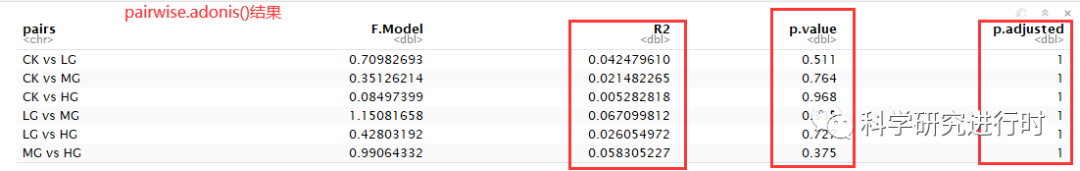
图7|PERMANOVA多重比较结果。
* p.adjusteds是校正后P值,此次使用的 bonferroni方法,其实还有很多校正方法可以选择,如FDR和BH等。P.adjusted值大于0.05,表明R2结果不具有统计学意义。
* 其他参数意义与整体检验一致
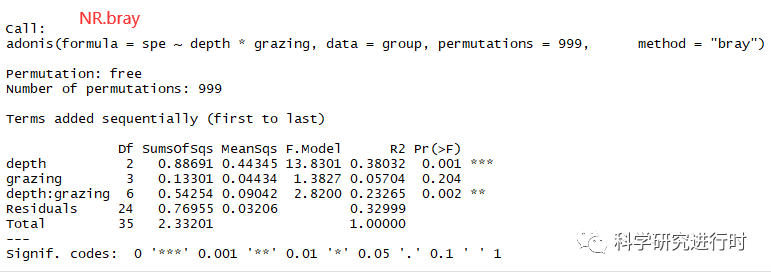
图8|多个分类因子交互作用分析结果。
* depth,grazing分别表示土壤深度或放牧对微生物群落结构差异的解释度
* depth:grazing,表示土壤深度与放牧两个分类变量对微生物群落结构差异解释度。
* 其它参数意义相同
3. ANOSIM-NMDS
ANOSIM(Analysis of similarities)是一种非参数相似性检验,可用于检验不同群落结构的相似性以及相似性是否显著,可以是基于分类变量,也可以来自排序或聚类分析得到的分类。
3.1 分析流程
# ANOSIM群落结构差异分析-bray-curits index
NRCK.anosim<-anosim(spe,group$depth,permutations = 999,distance = "bray")
NRCK.anosim
# distance.bray<-vegdist(spe,method = 'bray') #也可以使用vegdist()函数计算完距离之后再进行群落结构差异分析
# grazing.bray = anosim(distance.bray,group$depth,permutations = 999") # 先计算完距离之后,就不用写method参数
# 多重比较
pairwise.anosim <-function(x,factors, sim.method, p.adjust.m){
library(vegan)
co = as.matrix(combn(unique(factors),2))
pairs = c()
R = c()
p.value = c()
for(elem in 1:ncol(co)){
ad = anosim(x[factors %in%c(as.character(co[1,elem]),as.character(co[2,elem])),],
factors[factors %in%c(as.character(co[1,elem]),as.character(co[2,elem]))], permutations = 999,distance = "bray");
pairs = c(pairs,paste(co[1,elem],'vs',co[2,elem]));
R = c(R,ad$statistic);
p.value = c(p.value,ad$signif)
}
p.adjusted =p.adjust(p.value,method=p.adjust.m)
pairw.res = data.frame(pairs, R, p.value,p.adjusted)
return(pairw.res)
} # 不需要更改
pairwise.anosim(spe, group$grazing, sim.method="bray", p.adjust.m= "fdr") # 多重比较,使用FDR进行P值校正ANOSIM检验使用的是组内或组间距离的平均值,对样本量、数据异质性(方差不齐)很敏感,方差不齐最好不要使用。
笔记中还有语句“ANOSIM适用于样本在欧式平面中单个数据变化有重要意义的情况”,我已经忘记是在哪里看到的,是什么意思了,要是有大神知道什么意思,希望能后台发我一下。
3.2 结果解读

图9|ANOSIM分析结果。
* R∈[-1,1],表示群落相似性,R值越接近1表示组间差异大于组内差异,值越大表示组间差异越大。
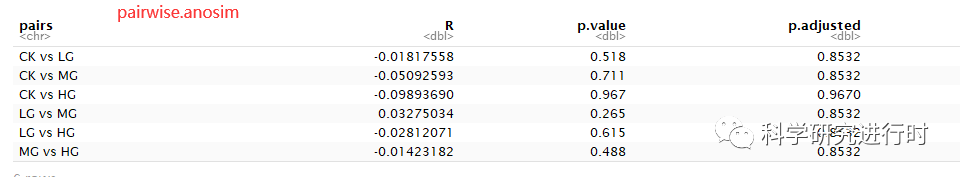
图10|ANOSIM多重比较结果。结果解读与整体比较一致。
4. MRPP
MRPP(多元响应置换分析)是一种与ANOSIM类似的分析组间群落结构差异是否显著的方法,不同的是它用的是群落的原始差异而不是rank。
4.1 分析流程
# MRPP群落结构差异分析-bray-curits index
NRCK.mrpp<-mrpp(spe,group$depth,permutations = 999,distance = "bray") # 也可以使用dist和vegdist计算群落距离。
NRCK.mrpp
# 多重比较
pairwise.mrpp <-function(x,factors, sim.method, p.adjust.m){
library(vegan)
co = as.matrix(combn(unique(factors),2))
pairs=c()
A = c()
ObservedDelta=c()
ExpectedDelta = c()
p.value=c()
for(elem in 1:ncol(co)){
ad = mrpp(x[factors %in%c(as.character(co[1,elem]),as.character(co[2,elem])),],
factors[factors %in%c(as.character(co[1,elem]),as.character(co[2,elem]))], permutations = 999,distance = sim.method);
pairs = c(pairs,paste(co[1,elem],'vs',co[2,elem]));
A = c(A,ad$A);
ObservedDelta = c(ObservedDelta,ad$E.delta)
ExpectedDelta = c(ExpectedDelta,ad$delta)
p.value=c(p.value, ad$Pvalue)
}
p.adjusted =p.adjust(p.value,method=p.adjust.m)
pairw.res = data.frame(pairs, A, ObservedDelta, ExpectedDelta, p.value, p.adjusted)
return(pairw.res)
} # 不需要更改
pairwise.mrpp(spe, group$depth, sim.method="bray", p.adjust.m= "fdr") # 多重比较4.2 结果解读
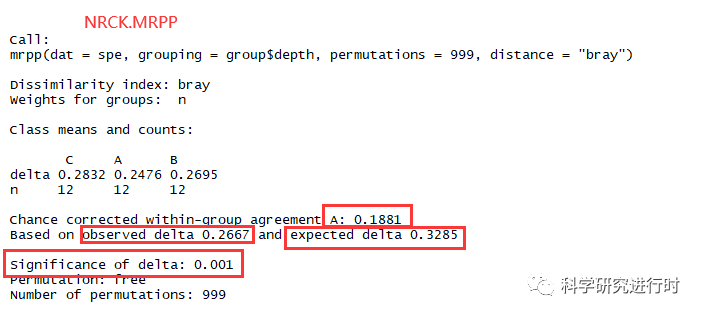
图11|MRPP整体分析结果。
* A=0.1881,A>0表示组间差异大于组内差异,A<0表示组间差异小于组内差异。
* observed delta=0.2667,值越小说明组内差异越小。
* expected delta=0.3285,值越小说明组间差异越小。
* significance of delta(P)=0.001,小于0.05说明结果有统计学意义。
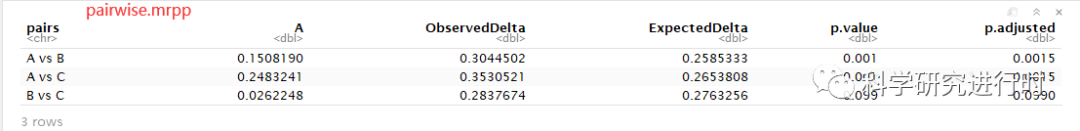
图12|MRPP多重比较结果。各值的意义与上面一致。
4. Mantel
上面三种群落结构分析方法都是基于分类变量进行的分析,而基于连续变量的群落结构分析使用Mantel检验和variation partition analysis(envfit()函数,VPA)。VPA在R绘图-RDA排序分析中已经讲过了,这里就只讲Mantel。
Mantel()函数用于对两个相异矩阵进行相关性分析,如分别对微生物组成数据和环境因子(C、N、P含量等)计算距离矩阵,然后进行环境因子变化与微生物群落结构间的相关性(或解释度),可以根据数据情况选择使用pearson、spearman或 kendall相关性指数。
4.1 分析流程
# 群落结构分析-Mantel
library(ecodist)
spe.dist<-vegdist(spe,method = 'bray')
soil.dist=vegdist(env,method = "euclidean") # 整体环境因子数据一起分析,环境因子矩阵一般用欧式距离
soil.mantel = vegan::mantel(spe.dist,soil.dist,permutations=999,method="pearson") # vegan::mantel(),使用vegan包中的mantel()函数
soil.mantel
#pH.dist=vegdist(env$pH,method = "euclidean") # 单个环境因子分析
#pH.mantel= vegan::mantel(spe.dist,pH.dist,permutations=999,method="pearson")
#pH.mantel
# 每个环境因子单独进行相关性分析
single.mantel <-function(spe,env, sim.method, correlation, p.adjust.m){
library(vegan)
co = colnames(env)
factor = c()
r = c()
p.value=c()
spe.dist<-vegdist(spe,method = sim.method)
for(elem in 1:length(co)){
env.dist = vegdist(env[elem], method = "euclidean")
ad = vegan::mantel(spe.dist, env.dist, permutations = 999,method=correlation);
factor = c(factor,co[elem]);
r = c(r,ad$statistic);
p.value=c(p.value, ad$signif)
}
p.adjusted =p.adjust(p.value,method=p.adjust.m)
pairw.res = data.frame(factor, r, p.value, p.adjusted)
return(pairw.res)
} # 不需更改
sin.mantel = single.mantel(spe, env, sim.method="bray", correlation = "pearson", p.adjust.m= "fdr") # 多重比较
sin.mantel
# 将结果输出到csv文件中
write.csv(sin.mantel,"sin.mantel.csv",row.names = FALSE)4.2 结果解读
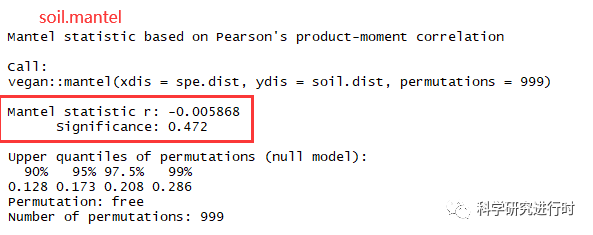
图13|Mantel整体环境因子分析结果。单个环境因子结果个参数意义一致。
* r, 微生物群落β-多样性距离矩阵与环境因子距离矩阵之间的相关性
* Significance(p),p<0.05表明环境因子与微生物群落距离矩阵的相关性有统计学意义。
* 所有环境因子一起计算距离矩阵时要注意,环境因子数不要超过样本数,envfit()函数也一样。
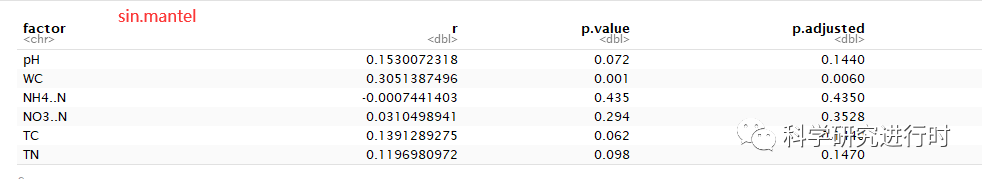
图14|Mantel 每个环境因子单独与群落距离矩阵的pearson相关性。参数意义与上面一致。
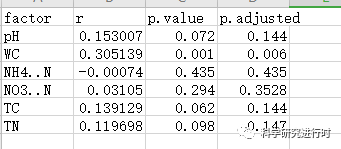
图15|sin.mantel.csv文件。
5. Mantel.partial
vegan包中还有一个mantel.partial()函数,当环境因子之间本身存在相关性时,可以先利用方差膨胀因子分析对环境因子做相关分析,然后将显著与其它环境因子相关的环境因子作为协变量,其它环境因子作为变量进行mantel.partial分析,也可以将感兴趣的环境因子作为变量,其它环境因子作为协变量进行分析。
# 群落结构分析-Mantel.partial
spe.dist<-vegdist(spe,method = 'bray')
spe.dist
main.dist=vegdist(env[1:3], method = "euclidean")
main.dist
covar.dist=vegdist(env[4:6], method = "euclidean")
covar.dist # 协变量
mantel.partial(spe.dist, main.dist, covar.dist, method = "pearson", permutations = 999)
图16|Mantel.partial分析结果。参数意义与上面一致。
根据环境因子相关性筛选用于mantel.partial分析协变量的方法,宏基因组公众号发的“微生物环境因子分析(RDA/db-RDA)-ggvegan”一文中有写,想用此方法的同学可以去看看,因为我不经常用这个功能所以这里就不写了。Mantel.partial分析的单个环境因子相关性的批量运行,大家可以参照mantel分析自己改代码进行分析,就当是逼着大家要读懂这些代码了 。
。
6. 参考资料
β-多样性算法
推荐阅读
微生物环境因子分析(RDA/db-RDA)-ggvegan包
R统计-PCA/PCoA/db-RDA/NMDS/CA/CCA/DCA等排序分析教程
R统计绘图-单、双、三因素重复测量方差分析[Translation]
R统计绘图-corrplot热图绘制细节调整2(更改变量可视化顺序、非相关性热图绘制、添加矩形框等)
我把原始数据、分析流程和结果都打包放在了百度网盘中了,大家也可以后台回复“群落结构差异分析教程”,直接将网盘链接发给大家,大家可以自己运行一篇。数据分析不仅要看,还得不停实操才行。希望大家在没有要用的时候就学起来,不然等急着要用的时候,时间就很紧张了。最最起码要做到,当自己要做某个分析时,知道要去哪里找笔记或者教程。

EcoEvoPhylo :主要分享微生物生态和phylogenomics的数据分析教程。
扫描左侧二维码,关注EcoEvoPhylo。让我们大家一起学习,互相交流,共同进步。
学术交流QQ群 | 438942456
学术交流微信群 | jingmorensheng412
加好友时,请备注姓名-单位-研究方向。
开放转载
公众号原创文章开放转载,在文末留言告知即可

旨在为数千万中国开发者提供一个无缝且高效的云端环境,以支持学习、使用和贡献开源项目。
更多推荐


 10
10 0
0- 0



 已为社区贡献7条内容
已为社区贡献7条内容

所有评论(0)